
Guidance for Industry on Providing Regulatory Information in Electronic Format
Harmonised Technical Guidance for
eCTD Submissions in the EU
Version 4.0
April 2016
Page 1 of 62

Table of Contents
Table of Contents ........................................................................................................................ 2
List of Tables ............................................................................................................................... 4
List of Figures .............................................................................................................................. 4
1. INTRODUCTION .............................................................................................................. 5
1.1 Glossary ...................................................................................................................... 6
2. GENERAL CONSIDERATIONS ...................................................................................... 7
2.1 Scope .......................................................................................................................... 7
2.2 Structure of Submissions ............................................................................................. 7
2.3 Transitional Arrangements........................................................................................... 8
2.4 Moving to eCTD Format from Paper or NeeS Type Applications ................................. 8
2.5 General Submission Considerations ............................................................................ 9
2.6 Correspondence ........................................................................................................ 10
2.7 Paper Requirements.................................................................................................. 10
2.8 Hardware ................................................................................................................... 10
2.9 General Technical eCTD Information ........................................................................ 10
2.10 Other Technical Information ...................................................................................... 16
2.11 Number of Media Requested ..................................................................................... 19
2.12 Technical Baseline Applications ................................................................................ 19
3. MODULE SPECIFIC INFORMATION ............................................................................ 23
3.1 General Information ................................................................................................... 23
3.2 Module 1 eCTD Envelope, Administrative Information and Prescribing Information Folder
23
3.3 Module 2 Overviews and Summaries Folder ............................................................. 27
3.4 Module 3 Quality Folder ............................................................................................ 27
3.5 Module 4 Nonclinical Study Reports Folder ............................................................... 27
3.6 Module 5 Clinical Study Reports Folder ..................................................................... 28
4. ADVICE ON SPECIFIC APPLICATION TYPES ............................................................ 29
4.1 New MA Applications ................................................................................................. 29
4.2 Variation Applications ................................................................................................ 30
4.3 Extension Submissions ............................................................................................. 32
4.4 Renewal Submissions ............................................................................................... 32
4.5 PSURs ...................................................................................................................... 33
4.6 Referrals .................................................................................................................... 34
4.7 Active Substance Master Files .................................................................................. 35
4.8 Vaccine Antigen Master Files .................................................................................... 35
4.9 Plasma Master Files (PMFs) and medicinal products containing PMFs ..................... 36
4.10 Applicant Initiated Withdrawals of the MA or certain strengths, dosage forms ........... 36
4.11 Applicant Withdrawal or Agency Rejections of Post-Authorisation Regulatory Activities36
4.12 Publication of Clinical Data for Medicinal Products .................................................... 37
4.13 Duplicate Applications and Informed Consent Applications ....................................... 37
A
NNEX 1: ECTD REFERENCE DOCUMENTS ........................................................................... 38
A
NNEX 2: GUIDANCE ON TEXT SEARCHABLE DOCUMENTS .................................................... 39
A2-1 General ..................................................................................................................... 39
A2-2 Documents that Must Always Be Text Searchable .................................................... 39
A2-3 Documents that Do Not Need to Be Text Searchable ................................................ 40
A2-4 Further Information .................................................................................................... 40
A
NNEX 3: GUIDANCE AND BEST PRACTICE ON THE STRUCTURE OF MODULE 3 ....................... 41
A3-1 Introduction ............................................................................................................... 41
Technical eCTD Guidance v4.0 Page 2 of 62

A3-2 General Principles ..................................................................................................... 42
A3-3 Module 3 XML Attributes in the eCTD ....................................................................... 44
A
NNEX 4 – MANAGEMENT OF PARALLEL VARIATIONS IN THE ECTD .......................................... 54
A4-1 Background ............................................................................................................... 54
A4-2 Business Challenge ................................................................................................... 54
A4-3 Best Practice ............................................................................................................. 54
A4-4 Description of Figures ................................................................................................ 55
Technical eCTD Guidance v4.0 Page 3 of 62

List of Tables
Table 1 : Example of a PSUSA ......................................................................................................... 12
Table 2 : Example of a Referral for CAPs ......................................................................................... 12
Table 3 : Example of a Referral for NAPs ......................................................................................... 12
Table 4 : Repeatable Elements and Attributes that Define Different Sections ................................... 13
Table 5 : Example for starting an eCTD with a baseline sequence ................................................... 19
Table 6 : Example for starting an eCTD with regulatory activity sequence ........................................ 20
Table 7 : Examples of Filenames and Leaf Titles for Response Documents ..................................... 26
Table 8 : MAA – Centralised Procedure ........................................................................................... 29
Table 9 : Centralised Procedure - Outside eCTD via EudraLink ....................................................... 30
Table 10 : New MAA – Decentralised Procedure .............................................................................. 30
Table 11 : Type II Variations – Centralised Procedure ...................................................................... 30
Table 12 : Type IA & IB Variations – Centralised Procedure ............................................................. 31
Table 13 : Type IB Variations with linguistic review - Centralised Procedure .................................... 31
Table 14 : Renewals ......................................................................................................................... 33
Table 15 : Centralised Procedure – Outside eCTD via EudraLink .................................................... 33
Table 16 (A3) : Advantages and disadvantages of eCTD application structures ............................... 42
List of Figures
Figure 1 : Sample of folder structure................................................................................................. 15
Figure 2 (A3) : Single Drug Substance, 2 Manufacturers with similar documentation, the few
site/manufacturer-specific documents are identified by the XML title (not by adding an additional XML
section): ........................................................................................................................................... 46
Figure 3 (A3) : Single Drug Substance, 2 Manufacturers with significant volume of different
documentation (one section for Acme Chemicals, another for Pharmasupply) ................................. 48
Figure 4 (A3) : Approach 1 – One 32p for all Strengths, any strength specific documents identified by
the XML title, not by adding an additional XML section ..................................................................... 50
Figure 5 (A3) : Approach 2 - Separate XML elements and documents for Strengths – significant
content differences, but Pharmaceutical Development only provided once in the folder structure and
referred to from the XML twice ......................................................................................................... 52
Figure 6 (A4): Description of elements ............................................................................................. 55
Figure 7 (A4) : Use of one document lifecycle .................................................................................. 55
Figure 8 (A4) : Use of separate document lifecycles with different title attributes .............................. 57
Figure 9 (A4) : Replacement of approved content by newly approved content, and deletion of
proposed content .............................................................................................................................. 57
Figure 10 (A4) : Progression on the 2nd Parallel Proposal ............................................................... 59
Figure 11 (A4) : Replacement of approved content by newly approved content, and deletion of
multiple proposed documents ........................................................................................................... 60
Technical eCTD Guidance v4.0 Page 4 of 62

1. INTRODUCTION
This guidance document is intended to assist pharmaceutical companies with the submission of regulatory
information in electronic Common Technical Document format (eCTD) to the National Competent Authorities
(hereafter referred to as NCAs) and the European Medicines Agency (hereafter referred to as EMA). The eCTD
format is regarded as the principal electronic submission format in EU for human medicinal products and is the
only electronic format that is accepted by the EMA (except for some specified procedures) and is stepwise
becoming mandatory within the Decentralised, Mutual Recognition Procedures and purely National Procedures
as well depending on national decisions. However, the Non eCTD electronic Submissions (NeeS) format is still
accepted within these procedures and for National Procedures and therefore a guidance document for NeeS
is
also published on the EMA eSubmission website.
This guidance was initially created by the TIGes Harmonisation Group, a sub-group of the Telematics
Implementation Group for electronic submissions (TIGes), and is now being maintained by the Human
Harmonisation Maintenance Group (HHMG), a sub-group of the eSubmission Change Management Board
(CMB).
It should be stressed that this guidance reflects the current situation and will be regularly updated in the light of
changes in national and/or European legislation together with further experience gained within NCAs and EMA
using information submitted in electronic format. If needed, there are also Q&A documents published in
between versions of this guidance as a response on change requests or new requirements to be addressed
(see EMA eSubmission website
)
This document consists of four parts: Introduction, General Considerations, Module Specific Information and
Advice on Specific Application Types together with associated annexes.
Technical eCTD Guidance v4.0 Page 5 of 62

1.1 Glossary
A brief glossary of terms (for the purpose of this document only) is indicated below:
Term
Definition
Applicant
A pharmaceutical company or its agent that is submitting information in
support of an application.
Applicant’s Information
Regulatory information submitted by an applicant for, or to maintain, a
marketing authorisation that falls within the scope of this guidance
document.
eCTD application or also
known as a dossier
A collection of electronic documents compiled by a pharmaceutical
company or its agent in compliance with European legislation and
guidelines in order to seek a marketing authorisation or any
amendments thereof. An eCTD application may comprise a number of
regulatory activities. In the EU an eCTD application may comprise
several dosage forms and strengths, all under one invented product
name. This is understood to be equivalent to a Global Marketing
Authorisation according to Art. 6 para 2 Dir. 2001/83/EC as amended.
Procedure
A Community registration procedure for the authorisation of medicinal
products in the European Community. There are 4 types of procedure
that operate within the EC – Centralised, Decentralised, Mutual
Recognition and National.
Regulatory Activity
A single sequence or a collection of sequences covering the start to
the end of a specific business process, e.g. an MA application or Type
II variation. To allow a more precise handling, the regulatory activity will
be classified using a controlled vocabulary (submission type or
regulatory activity type) and a free text field for a short narrative
description.
Sequence
A single set of information and / or electronic documents submitted at
one particular time by the applicant as a part of, or the complete
application .Any collection of content assembled in accordance with the
eCTD specification (ICH and EU) will be described using metadata as
defined by the EU envelope. Sequences may be related to one another
within one regulatory activity. The related sequence number should
always be stated. In case of activities with only one sequence the same
sequence number will be used.
Submission Type
The submission type describes the regulatory activity to which the
content will be submitted.
Submission Unit Type
The submission unit type element of the envelope metadata set
describes the content at a lower level (a “sub-activity”) which is
submitted in relation to a defined regulatory activity such as the initial
submission, the applicant response to validation issues or list of
questions or any other additional information.
Technical eCTD Guidance v4.0 Page 6 of 62

2. GENERAL CONSIDERATIONS
2.1 Scope
2.1.1 Types of Product
This guidance covers the submission of electronic regulatory information for all human medicinal products
falling within the competence of NCAs in the EEA as well as the EMA. This includes but is not limited to
prescription, over the counter medicines, innovative and generic product submissions. The product types
include for example small molecules, biotech products, herbals, vaccines, homeopathics or blood products.
2.1.2 Types of Submission
This guidance applies to all submissions related to the authorisation and maintenance of medicinal products,
including new marketing authorisations, variations, renewals, PSURs (including PSUR Single Assessment
PSUSA), active substance master files (ASMF), Plasma Master Files (PMF) and withdrawals, submission of
redacted clinical trial reports as well as any kind of paediatric submissions and referral related or post
authorisation measures related submissions. For variations, PSUSAs, ASMF and PMF there are also specific
guidance documents (see references in Part 4
).
2.1.3 Types of Submission Units
This allows sequences to be grouped together that make up a Regulatory Activity. The submission unit will
describe the sub-activity within a Regulatory Activity, such as initial, validation-response, response and closing
in case of a new MAA.
The submission unit ‘additional-info’ should be used for additional information (which could include, for
example, missing files), and should only be used when ‘validation-response’ or ‘response’ is not suitable. The
submission type ‘corrigendum’ should only be used in exceptional circumstances in the CP to correct
information, typically for product information, after the Regulatory Activity has concluded.
2.1.4 Types of Procedures
This guidance covers applications made in any of the applicable Community procedures (National, Mutual
Recognition, Decentralised and Centralised). For submissions within MRP and DCP, please refer to the
specific CMDh guidance ‘
Requirements on submissions for New Applications within MRP, DCP or National
procedures’
2.1.5 Exceptions
This guidance does not apply to the electronic submission of pre-marketing authorisation (MA) information such
as scientific advice, clinical trial applications, orphan drug designations, PIP submissions and related
submission correspondence as well as dossier content explicitly excluded from the commonly maintained
electronic dossier. These exceptions may be subject to change in the future. (Please refer to the EMA website
and to CMDh website on eSubmission for the BPG and Q&As for further exceptions.)
2.2 Structure of Submissions
This document provides guidance on how to organise application information for electronic submission using
the eCTD specifications. Guidance on the detailed information to be included is described in the
Common
Technical Document (CTD), and relevant ICH and EU Q&A documents.
The structure and organisation of an eCTD submission is defined by the following standards:
• ICH M2 eCTD Specification
• EU Module 1 Specification
• Relevant ICH and EU Q&A docs
Annex 1
contains links to the currently approved version of these documents.
Technical eCTD Guidance v4.0 Page 7 of 62

Typically, an eCTD application will cover all dosage forms and strengths of a product. In the centralised
procedure, this will be equivalent to all dosage forms and strengths covered by an EMA application number
(e.g. EMEA/H/C/000123). In MRP/DCP, a single eCTD application should preferably be used for the procedure.
However if an applicant decides not to apply for all strengths and dosage forms in every member state in the
procedure, the possibility of having one eCTD application per strength/dosage form should be considered.
Applicants should carefully consider what an eCTD application should cover before submitting the first
sequence, as the choice could have implications for workload for the lifespan of the product. For example, if the
applicant decides to have one eCTD per strength or dosage form, it is expected that each of these eCTD
applications will be maintained individually, such that submission of a single sequence that covers more than
one strength or dosage form will no longer be possible if very good reasons are not presented for a change
over. In these rare cases, please contact the NCA/RMS/EMA concerned at an early planning stage.
For further details on the pros and cons of the different approaches to dossier structure,
see Table 16 (A3):
Advantages and disadvantages of eCTD application structures.
Please check for specific NCA guidance when preparing national eCTDs. However, note that the selection of
separate lifecycles for national (MRP/DCP/NP) products will mean in practice that submissions for EU PSUR
Single Assessment must be made for each product separately in accordance with the existing dossier structure
(for details see Section 4.5
).
2.3 Transitional Arrangements
The specifications mentioned in Section 2.2 above will change over time and are likely to affect both eCTD
building tools and the applicant’s internal business processes as well as the agencies review tools and
processes. Once a new specification has been agreed and endorsed by the appropriate EU body, eCTD
building tools will need to be updated. Specific transitional guidance will be provided on each occasion that the
ICH and/or EU specifications are updated.
Please note that it should not be necessary to reformat and resubmit previously submitted applications to
reflect such changes.
2.4 Moving to eCTD Format from Paper or NeeS Type Applications
Changing format from paper or NeeS to eCTD can be done at a start of any regulatory activity such as an
extension, a renewal or a variation, ideally when no other regulatory activities are on-going for that product in
another format. A baseline submission is recommended at the time of changing to eCTD (see Section 2.12
.) to
give the agencies access to all or at least part of the previously submitted documentation within the eCTD
lifecycle. When the eCTD lifecycle is initiated and accepted by the authorities, all further submissions related to
that product dossier should from that day be submitted in eCTD format. This may also include submissions
concerning other ongoing regulatory activities related to that eCTD application (e.g. responses to questions to
ongoing variations), in which case, it will obviously not be possible to use the related sequence attribute
correctly since the start of the regulatory activity is not present as an eCTD sequence to refer to and therefore
the validation criterion 14 BP2 will not be met. This should be reflected in the cover letter.
If the dossier has already been provided in NeeS format, the applicant should submit the new data in eCTD
format starting the lifecycle in accordance with eCTD specifications. The first submission in eCTD format will
normally be sequence 0000, even if sequential numbers were used for the NeeS format. For clarity, the cover
letter should always explicitly state that the submission involves a switch to eCTD format. As the documents
already exist in an electronic format, it would be preferable to first re-send the currently valid documents,
especially module 3, as a baseline eCTD dossier in sequence number 0000 and then the first new regulatory
activity as 0001. Please see Section 2.12
for further information on the content of baseline applications and the
acceptability of scanned documents.
If an applicant has been submitting applications in eCTD format to some agencies within MRP or DCP when
paper or NeeS were still requested by some other NCAs – in the context of the switch to eCTD format of these
remaining paper/NeeS-based agencies, it would be of help if applicants would submit all former eCTD
sequences in connection to a new regulatory activity for that product. The “old” eCTD sequences should be
Technical eCTD Guidance v4.0 Page 8 of 62

provided together with this new eCTD sequence and it should be clearly stated in the cover letter to the
concerned NCAs that the “old” sequences have the same content as formerly submitted paper or NeeS format
documents. When submitting earlier sequences to other agencies, no changes to envelopes or metadata is
required, it is accepted that the envelopes might not be entirely correct for agencies receiving a sequence
previously submitted to another agency. Any historical sequences should not be technically validated by the
agencies receiving them for the first time, for details see the CMDh guidance ‘
Requirements on submissions for
New Applications within MRP, DCP or National procedures’. However, if there are problems with loading or
reading the “old” files, the applicant should assist in solving the technical problems on the sequences to
facilitate their use in the “new” NCA, for example due to mistakes in transmission or creating the submission or
problems with the XML, which can be resolved without affecting future lifecycle.
In any case, a tracking table is essential to understand the sequencing of your eCTD submission (please refer
to Section 3.2.3
).
Where the change from paper or NeeS to eCTD format for a product dossier is planned to be done in
connection to a repeat use procedure (i.e. for the complete dossier), the change of format should first be made
in the RMS and the “original” CMSs by submitting the current dossier as a so called baseline dossier (see
Section 2.12
), before the start of the repeat use procedure in the new CMSs.
2.5 General Submission Considerations
2.5.1 Document Granularity
Submissions are a collection of documents and each document should be provided as a separate file. The
detailed structure of the eCTD should conform to the ICH Granularity Document
and EU M1 specification.
Careful consideration is needed when deciding the level of Module 3 granularity (please refer to Annex 3,
Section 3.1)
2.5.2 File Naming
The eCTD file naming conventions described in the ICH M2 eCTD Specification and EU Module 1 Specification
are highly recommended, as best practice. If an applicant wishes to submit multiple files in one section, where
only one highly recommended name is available, this can be achieved using a suffix to the filename, such as
using the file name-var.pdf convention as described in the EU Module 1 Specification (e.g. pharmaceutical-
development-container.pdf).
The variable part of the name must not contain “illegal” characters.
File names, including the extension, must not exceed 64 characters. Also folder names must not exceed 64
characters and the total file folder path length must not exceed 180 characters. Counting starts from the first
digit of the sequence number in the sequence number folder name.
For further guidance on file naming, please refer to the “File-Folder Structure & Names” work sheet included in
the current validation criteria
2.5.3 Placement of Documents
Guidance on the placement of documents within the eCTD structure for particular submission types can be
found in the EU-CTD Notice to Applicants and/or in the EMA post-authorisation guidance
for centralised
applications.
In the submission structure, leaves should typically be placed at the lowest level of the CTD structure, although
there are some exceptions to this guidance, for example, in 32p4-contr-excip, where the files excipients.pdf,
excipients-human-animal-var.pdf or novel-excipients-var.pdf can be placed alongside folders containing details
of other excipients. The lowest levels of the CTD structure (including node-extensions) must contain at least
one leaf, although this should normally be managed automatically by the eCTD building tool.
Technical eCTD Guidance v4.0 Page 9 of 62

Every leaf must have a value for the ‘title’ attribute.
2.6 Correspondence
The eCTD is designed to ensure that users have a current view of the information submitted in the appropriate
place in the dossier at all times. Therefore, formal responses to questions should always be submitted in eCTD
format, as well as any correspondence that relates directly to the content of the dossier.
In addition to the eCTD application, information may need to be exchanged to assist the processing or handling
of the application. If this correspondence is not directly relevant to the application dossier then it should not be
included in the eCTD. This is because the eCTD exchange is currently one way only, from applicant to
authority. Other correspondence should be exchanged outside the eCTD via the usual electronic means (email,
EudraLink etc.).
2.7 Paper Requirements
Some NCAs may still require paper copies of some documents in addition to the eCTD; refer to the CMDh
guidance ‘Requirements on submissions for New Applications within MRP, DCP or National procedures
’ for
further details.
2.8 Hardware
NCAs and the EMA will not accept any hardware (laptops, desktops, external hard drives etc.) from applicants
in connection with the submission of information in electronic format. The electronic information should be
directly readable and usable on NCAs and EMA hardware and software.
2.9 General Technical eCTD Information
2.9.1 Identifier for Applications
To help enable archiving the sequence with the correct dossier (eCTD lifecycle), applicants must state a unique
identification of the dossier in the form of a UUID. The identifier must be defined with the first sequence of a
new dossier (or the first sequence using EU M1 version 3.0 or later), and it must be consistent throughout the
eCTD lifecycle. The identifier must be changed if – and only if – the eCTD lifecycle is restarted. Examples of
this are addressed in Section 2.12.3
.
The UUID is a 32 digit (8+4+4+4+12) hexadecimal string, for example: “123e4567-e89b-12d3-a456-
426655440000”. When defining a UUID the string should be decided at random. The string should contain no
information referring to any other metadata. This is to ensure consistency even if the relevant metadata should
change. The chance of two different dossiers having the same UUID is next to none if the UUIDs are truly
defined at random.
2.9.2 File Formats
In general terms the majority of documents included in electronic submissions should be in PDF format (see
next section on the use of PDF file versions). Files that might be requested by NCAs or the EMA in MS Word
format should not be included in the eCTD structure (refer to Section 2.9.10
).
Further detailed guidance on file formats can be found in the ICH eCTD specification document and EU Module 1
specification.
Technical eCTD Guidance v4.0 Page 10 of 62

2.9.3 Portable Document Format (PDF)
Portable Document Format (PDF) is an ISO standard (ISO 19005-1:2005 Document management – Electronic
document file format for long-term preservation – Part 1: Use of PDF 1.4 (PDF/A-1) and ISO 32000-1:2008
Document management – Portable document format – Part 1: PDF 1.7). Although created by Adobe Systems
Incorporated there are several alternative suppliers of PDF software. Applicants need to check that their PDF
documents meet the following key requirements:
• Files should be legible with Acrobat Reader, version 5.0 or higher.
• PDF version 1.4, 1.5, 1.6 or 1.7 should normally be used, except where there is an agency specific
requirement for another version for example for application forms.
• PDF 1.3 or earlier versions are not acceptable for technical reasons. No exceptions will be made. For
example, if a literature reference is received in PDF 1.3 or earlier, then the applicant must convert it to PDF
1.4, 1.5, 1.6 or 1.7, either electronically or by scanning. .
• Documents generated by the applicant should be created from electronic source documents and not from
scanned material. Where access to the source electronic file is unavailable please refer to Annex 2
.
Additionally, the following requirements should be taken into consideration:
• Different requirements apply in the EU to NCAs and the EMA for signatures on application forms and cover
letters. – For details refer to Section 2.10.4
. Make sure that scanned cover letters are text searchable.
• The fonts used in the PDF should be embedded in the PDF where possible.
• Rendition to PDF should preferably create documents which are ”tagged”.
• Use 'Fast Web View' to ensure optimum performance of the review system. Due to technical constraints for
eAF documents the fast web view cannot be enabled. Therefore, the BP warning for eAF can be ignored.
.
Additional details on PDF, including those relating to the good presentation of tables, can be found in the
ICH
eCTD Specification, Appendix 7. Refer also to the ICH M2 recommendations.
2.9.4 Sequence Numbers
Sequence numbers are used to differentiate between different submissions of the same application over the
lifecycle of the product. The review tools being used by most NCAs and the EMA are able to handle sequences
submitted out of numerical order, i.e. 0003 submitted after 0004. This can occur when the preparation of a
sequence is delayed. However, it is recommended that sequence numbers should normally follow the order of
submission of the sequences, as sometimes a higher sequence is technically related to the previous – not yet
submitted – sequence which will result in technical invalidation. A Sequence Tracking Table should always be
included in section 1.0 in every submission within MRP/DCP as a separate PDF file named cc-tracking-var.pdf.
For details see Section 3.2.3.2
.
The initial eCTD lifecycle submission should normally have a sequence number of 0000, even if sequential
numbers were already used for a NeeS format of the same product. If applicants consider that there are good
reasons to use another number they should explain this in the cover letter.
When additional information is submitted in response to questions or when information in a previously
submitted sequence is modified in any way, the sequence number of the submission will advance accordingly,
e.g. 0001, 0002, etc. Only in the case of a technically invalid submission, at request from or agreement with the
EMA (CP) or an NCA (MRP/DCP/NP), a sequence can be replaced with another using the same number (e.g.
the initial sequence “0000” will be replaced by another “0000”). The new sequence should be sent to all
concerned authorities. No new documents may be included in these cases, but only technical problems should
be fixed. The reason for resending the same sequence must be clearly stated in a comment in the delivery file
(CESP, working documents or separate note in case of CD/DVD submissions). For submissions to the EMA it
is mandatory to use the eSubmission Gateway / Web Client and no additional comment is required. If the eCTD
needs to be updated due to content/regulatory validation, any revised content should be provided with a new
sequence number and the changes clarified in the cover letter.
2.9.5 Related Sequence
All submissions should contain a value for “related sequence”. If the submission unit type is ‘initial’ or ‘reformat'
then the related-sequence attribute must have a value equal to the current sequence.
Technical eCTD Guidance v4.0 Page 11 of 62

If the submission unit type is another one than ‘initial’ or ‘reformat' then the entry for related sequence should
be populated with the number of the sequence that started the regulatory activity.
Table 1 : Example of a PSUSA
Sequence
number
Submission Description
Submission Type
Related
Sequence
Submission
Unit
0008
PSUR single assessment
procedure
psusa
0008
initial
0009
Validation update
psusa
0008
validation-
response
0010
Comments on Assessment
Report
psusa
0008
response
Table 2 : Example of a Referral for CAPs
Sequence
number
Submission Description
Submission Type
Related
Sequence
Submission
Unit
0008
Responses to LoQ
referral
none
initial
0009
Comments on Assessment
Report
referral
0008
validation-
response
0010
Responses to LoOI
referral
0008
response
Table 3 : Example of a Referral for NAPs
Sequence
number
Submission Description
Submission Type
Related
Sequence
Submission
Unit
0008
Responses to LoQ
referral
none
initial
0009
Comments on Assessment
Report
referral
0008
validation-
response
0010
Responses to LoOI
referral
0008
response
It is generally expected that there is usually just one Related Sequence, but, there are some occasions where
more than one Related Sequence should be provided as for example:
1) When there are two PAM related sequences (sequence 0005 and sequence 0006) and a single
response (sequence 0007) is produced that relates to both PAM related sequences.
2) When there are two parallel variations (sequence 0002 and sequence 0003) and there is a sequence
(0004) that brings the label up to date by including the changes made in both variations.
On these occasions multiple related sequences are used, but if a subsequent sequence relates to only one of
the original regulatory activities, then only the related sequence for that particular regulatory activity should be
used.
If the related sequences refer to both a single and grouped variation, the metadata should state ‘grouped’ as
being the highest level of regulatory activity.
Further examples are provided in the EU eCTD M1 Specification document.
Technical eCTD Guidance v4.0 Page 12 of 62

2.9.6 Leaf Lifecycle Operation Attributes
The leaf lifecycle operation attributes, as stated in the eCTD Specifications, are ‘new’, ‘append’, ‘replace’ and
‘delete’. However, in the EU, it is recommended that applicants avoid the use of ‘append’ due to the potential
for increased lifecycle complexity.
Lifecycle operations where the leaf targeted by the modified file is in a different CTD section are not allowed.
This applies across all of the modules of the CTD and is not specific to either the regional or ICH Modules.
Where elements are repeatable, both the element itself and the attributes that define a different position in the
table of contents must be identical when modifying a leaf element in a previous sequence. Where node
extensions are allowed, the node extension title element should be identical when modifying a leaf element in a
previous sequence. However, it is acceptable occasionally if inconsistencies have been introduced in the past.
In those cases, it is recommended to use the most recent version of the title attribute.
Table 4 : Repeatable Elements and Attributes that Define Different Sections
Element
Attributes also identifying a section
Module 1
m1-0-cover
country
m1-2-form
country
m1-3-1-spc-label-pl
country
xml:lang
type
m1-3-2-mockup
country
m1-3-3-specimen
country
m1-3-4-consultation
country
m1-3-5-approved
country
m1-responses
country
m1-additional-data
country
Module 2
m2-3-s-drug-substance
substance
manufacturer
m2-3-p-drug-product
product-name
dosageform
manufacturer
m2-7-3-summary-of-clinical-efficacy
indication
Module 3
m3-2-s-drug-substance
substance
manufacturer
m3-2-p-drug-product
product-name
dosageform
manufacturer
Module 5
m5-3-5-reports-of-efficacy-and-safety-studies
indication
The only exception to this is the change in agency name at the EMA; leaves with the specific country attribute
‘ema’ or ‘emea’ should be considered equivalent and lifecycle must be allowed between them.
Note, in the eCTD XML, sections, leaves and node extensions may have other attributes, such as xml:lang,
font-library, version, keywords, ID etc. These attributes, if supplied, do not result in a new logical section in the
eCTD table of contents and therefore lifecycle between leaves where these attributes are not identical is
allowed; only the attributes in the table above define different eCTD sections.
Technical eCTD Guidance v4.0 Page 13 of 62

Examples:
Pass/Fail
• A leaf to be submitted in m4-2-2-5 in Sequence 0012 cannot replace/delete a leaf submitted in m4-2-
2-2 of Sequence 0010.
• A leaf in m1-responses cannot modify content in m1-0-cover (different section)
• A leaf in m3-2-s-drug-substance [manufacturer: abcd] [substance: xyz] cannot modify content in m3-
2-s-drug-substance [manufacturer: other] [substance: xyz], or any section other than the specific
manufacturer ‘abcd’ and substance ‘xyz’ section.
• A leaf in m1-0-cover with the specific attribute ‘uk’ cannot modify a leaf in m1-0-cover with a specific
attribute of anything other than ‘uk’.
• A leaf in m1-3-pi with the specific attributes <pi-doc xml:lang="en" type="combined" country="ema">
cannot modify a leaf with different attributes, such as <pi-doc xml:lang="fr" type="other"
country="ema">.
Best Practice Criteria
• A leaf in a node extension should only be modified by a leaf in the same equivalent node extension in
a subsequent sequence. For example, a leaf in a node extension in m5-3-5-1 of sequence 0000 with
a <title> attribute ‘Study 1234’ should not be modified by a leaf in a different node extension with a
<title> attribute ‘Study 1234 – update’ in sequence 0001 – the append/replace/delete leaf ideally
needs to be in an identical node extension in 0001 (‘Study 1234’ in the same location, m5-3-5-1).
If the applicant places content in the wrong CTD section and needs to correct this (either upon request from the
agency receiving the eCTD or because they wish to correct the mistake) then the way to do this is to create two
leaf elements in a subsequent sequence. The first leaf will use a “delete“ operation to remove from view the
incorrectly placed content. The second leaf will usually use a “new” leaf operation to locate the content in the
correct CTD section. The file does not need to be resubmitted, the “new” leaf can use the xlink:href attribute to
point to the originally submitted content in the earlier sequence.
However, applicants cannot submit a dossier using the old specification and DTD. Therefore, deleting the
content in the old section will involve lifecycle from the changed section (for example, m1-additional-data)
deleting content in the equivalent section with the original name (in this example, m1-additional). This may not
be possible in all eCTD building tools, and if so, applicants are advised to leave the original content as it is, but
to start providing new or replacement content in the revised section.
Note: Lifecycle operations across eCTD applications are not allowed.
2.9.7 Bookmarks and Hypertext Links
Navigation through an electronic submission is greatly enhanced by the appropriate use of bookmarks and
hypertext links. ICH Q&A number 53 states, “It is expected that any document that has a Table of Contents
(TOC) will have bookmarks (see the eCTD specification for details). Documents without TOCs should have
bookmarks included where it aids in the navigation around the document content. For example, a 4 page
document summarising findings could require bookmarks to aid navigation. However, a 300 page file
containing a single data listing might not require bookmarks as there is no further internal structure. Please
consult regional guidance documents for further details.”
In general terms, bookmarks and hyperlinks should be used to aid navigation. The overuse of hyperlinks may
confuse rather than help assessors and may cause problems later in lifecycle management. However,
hyperlinks back to previously submitted documents are welcome as well if pointing to the correct location.
Additional details on creating bookmarks and hypertext links in PDF documents can be found in the
ICH eCTD
Specification, Appendix 7.
With the current version of the eCTD specification, it is not possible to cross refer from one eCTD application to
another.
Technical eCTD Guidance v4.0 Page 14 of 62

2.9.8 Node Extensions
Node extensions may be used where additional navigation in the XML backbone is required. The primary place
where they may be used is in Module 5 where a node extension for each study may be useful to group together
the multiple leaves that make up the study and its modular appendices. Also, it could be useful to differentiate
reports associated with a different dosing regimen for the same indication. For Module 4 where there are multi-
file reports, node extensions can also be useful, or in Module 1, for differentiating different responses in m1-
responses – for further details see Section 3.2.6
. Currently, there is no provision for additional folders in m1-
responses. Therefore, the use of an additional folder in combination with a node extension is not allowed.
However, the use of node extensions should be limited to those areas where it is critical and consideration
should be given regarding the impact of the view for the reviewer since the inconsistent use of node extensions
can lead to unanticipated effects in the cumulative view of a submission.
When node-extensions are used the ‘title’ attribute in the XML backbone must have a value.
2.9.9 Extensible Mark-up Language (XML)
XML is the format for the backbone files for the eCTD. Details on XML can be found in the ICH eCTD
Specification Document, Appendix 7. Initiatives on the use of XML structured information are supported by
NCAs and the EMA for electronic application forms (eAFs). Please refer to EMA eSubmission website for
further details.
2.9.10 Other File Formats
Other file formats such as MS Word formats may be required by some NCAs or the EMA in addition to the PDF
requirement of the eCTD, especially for the provision of product information documents or the Module 2
documents. Please refer to the CMDh website
for further details on NCAs requirements.
The files referred to above should not be added as leaf elements within the eCTD structure. When submitted
with an eCTD, they should always be provided in a separate folder called “xxxx-workingdocuments” on the
same submission zip package or on the same CD/DVD containing the eCTD, where the number (xxxx)
matches the number of the eCTD sequence being submitted.
For PMF certification submissions the ePMF should be provided within the working documents folder. The
folder should be called “xxxx-workingdocuments” as for all other documents. For more information please refer
to the guidance on PMF eCTD Guidance document
.
If working documents for more than one NCA are submitted on the same submission, sub folders with the
country code should be used.
Figure 1 : Sample of folder structure
For information on translations being provided outside the eCTD, refer to Section 3.2.5
If, at any stage in a procedure, an e-mail or EudraLink message is used to send information, this does not
change the format requirement. The subject line of the message should always include as a minimum the
product name and procedure number for identification purposes.
The EMA does not accept submissions sent by email or EudraLink.
Technical eCTD Guidance v4.0 Page 15 of 62

2.9.11 Technical Validation of eCTD Submissions
The technical validation of an eCTD is a separate activity to the content validation of a submission and takes
place irrespective of the type of the submission. NCAs and the EMA have adopted a
common set of technical
validation criteria against which all eCTDs can be checked using eCTD review and validation tools. It is strongly
recommended that all sequences are checked and found technically valid according to the published validation
criteria.
Two categories of validation rules apply: “Pass/Fail”, and “Best Practice”:
Pass/Fail Criteria
These are a set of technical validation criteria that can either be passed or failed. eCTDs that fail to meet
one or more of these criteria will be returned to the applicant for correction and resubmission of the same
sequence number. All Centralised Procedure (CP) eCTD submissions via the eSubmission Gateway / Web
Client to the EMA are automatically run through a full technical eCTD validation and an automated ‘success’
or ‘failure’ acknowledgement is sent to the applicant/MAH. eCTD submissions for PSURs authorised via
MRP/DCP/NP are checked using a ‘tolerant’ eCTD validation checking only the structure of the eCTD
submission. This means that even if your submission has been successfully submitted via the eSubmission
Gateway/Web Client, a technical validation issue can be found by an NCA at a later stage but will be
coordinated by the EMA in that case.
Best Practice Criteria
These are validation criteria that it is considered good practice to ensure are correct in the submitted eCTD.
The applicant should make every effort to address these areas before the eCTD is submitted. eCTDs that
fail to meet one or more of these criteria will still be accepted by the receiving agency/agencies during
technical validation and it is possible that agencies may not even check these criteria during technical
validation.
Note: These criteria cannot test the correctness of the metadata. Therefore, applicants need to make
sure that all metadata are filled in correctly.
Errors found during the content validation including misleading information in the cover letter or an application
form to be corrected for e.g. one or a few member states should be resolved through the submission of a new
eCTD sequence using the next sequence number. These errors must never be resolved by resubmitting an
existing sequence by re-using the same sequence number.
If historical sequences that have already been submitted in another MS in the EU are supplied to a new NCA,
the receiving NCA should not technically validate these sequences, as they have already been accepted when
originally submitted. This could be the case where, for example, in repeat use, switching from parallel national
to comprehensive model, supply of eCTD sequences to an NCA where this same submission had been
formerly submitted in NeeS or paper format but in eCTD format to other NCAs. However, if there are problems
with loading or reading the newly submitted files, the applicant should assist in solving the technical problems
on the sequences to facilitate their use in the “new” NCA.
2.10 Other Technical Information
2.10.1 Security Issues
The physical security of the submission during transportation/ transmission is the responsibility of the applicant.
Once received by NCAs and the EMA, security and submission integrity is the sole responsibility of NCAs and
the EMA.
2.10.2 Security Settings
Submission or file level security is not permitted. If one-time security settings or password protection of
electronic submissions are used this could constitute grounds for the rejection of the submission.
Technical eCTD Guidance v4.0 Page 16 of 62

There must be no security setting to open any individual file. This includes passwords, certificate security,
adobe policy server settings, etc. There must be no further security settings applied to any individual file
(except for files in Modules 3.3, 4.3 and 5.4). For example, in Adobe Acrobat, all "restrictions" should be
"allowed" when viewing the Document Preferences > Security settings>.
2.10.3 Protection against Malware
The applicant is responsible for checking the submission for malware such as viruses. Checking should be
performed with an up-to-date virus checker. After receipt at NCAs and the EMA, a similar internal virus check
will be performed. If a virus is detected it will constitute grounds for rejection of the submission.
2.10.4 Signatures
Electronic signatures are currently accepted in the EU as being legally equivalent to handwritten signatures
(Directive 1999/93/EC). Some NCAs and EMA already have this capability, or are developing it.
The NCAs have different requirements for wet signatures or scanned signatures although a signature is not
always required - refer to the CMDh guidance ‘
Requirements on submissions for New Applications within MRP,
DCP or National procedures’ for more details. In regard to electronic submissions to the EMA see for details on
the eSubmission Gateway and on eSignatures.
2.10.5 Procedure for Sending Electronic Information
There are different ways of submitting electronic dossiers to NCAs and the EMA, including portals, CD-R and
DVD-R, and EudraLink/e-mail, if accepted by agencies. See CMDh website
for further details on NCA
requirements, and the EMA website for details on the centralised procedure. Normally, only one way should be
used, to avoid sending multiple copies of the same submission to the authority. If an additional transmission is
requested by an agency (for example, a sequence is submitted via EudraLink and followed up with another
copy of the same sequence through a different channel), then this should be explained with a note or hard copy
letter such that the receiving agency can easily identify that it is a re-submission. The EMA does not accept any
transmissions on CDs / DVDs or Eudralink.
If required any necessary paper documentation should be provided at the same time as the electronic
submission, see CMDh website
for further details. Submissions that are sent to the NCAs in split packages (ie.
cover letter without a CD/DVD), or on a separate date in duplicates, might be processed with a delay.
Authorities will not accept any hardware (laptops, desktops, separate hard drives, etc.) from applicants in
connection with the submission of information in electronic format. The electronic information or eCTD should
be directly readable and usable on the authority’s hardware (e.g. CD/DVD drive) using its own software.
2.10.5.1 Portals
For the submission of applications, it is strongly recommended to use portals instead of CD/DVD wherever
possible.
There are two European portals in use for regulatory submissions. The Common European Submission
Platform (CESP) can be used to send national or MRP/DCP submissions to majority of NCA(s) - please refer to
the CESP website
for further details. In addition, CESP can be used for submission of human centralised
procedure dossiers to many NCAs for those submission types that are not available via the Common
Repository. The EMA eSubmission Gateway/Web Client must be used for submissions to the EMA in the
centralised procedure and from 13 June 2016 as announced by the EMA Management Board, for all PSUR
submissions. For further details see the
eSubmission website and the eSubmission Roadmap.
There are also national portals in some countries, see CMDh website
and individual NCA websites for further
details.
2.10.5.2 CD / DVD
The current standard to burn CDs / DVDs is Universal Disk Format (UDF
), which has replaced the former ISO
standard 9660. Zipped files should not be used when sending CDs or DVDs.
Technical eCTD Guidance v4.0 Page 17 of 62

Applicants should provide the electronic information on the smallest number of discs possible, taking into
consideration the size of the submission.
If an individual eCTD submission is of such a size as to span several CDs, the provision of a DVD is
recommended. However, if CD-R must be used, when large applications are submitted it is inevitable that the
application will necessarily span multiple CDs. Where possible, individual modules should not be split over
multiple CDs (e.g. if possible, a single CD should contain Module 1, Module 2, if too large to fit on the same CD
should then go onto the next CD even if this requires CD 1 not to be filled to capacity and so on). If, in the case
of larger modules, where a split over multiple CDs is inevitably necessary, subfolders should be distributed in
sequence, and these subfolders should not be split between CDs, even if this requires a CD to be sent not full
to capacity.
It is the choice of the applicant if a separate CD/DVD is provided for each new sequence or if several
sequences (e.g. concerning several variations) for the same medicinal product (same eCTD) is provided on the
same CD/DVD. This should be clearly described in the cover letter and indicated on the disc label.
It is, however not recommended to include previously submitted sequences to the same agency on a CD that
contains a new eCTD sequence.
The EMA does not accept CDs / DVDs for Centralised Procedure submissions or for Referral submissions.
2.10.5.3 EudraLink / e-Mail (where applicable)
EudraLink and e-mail are not recommended submission channels for eCTD submissions and in most cases,
eCTD sequences should not be sent by EudraLink or e-mail. Details on the dossier requirements in National
Competent Authorities are available on the CMDh website
. Also, normally only one route for submissions
should be used (CESP, EudraLink/e-mail or CD/DVD). Please note that EMA does not accept eCTD
sequences via EudraLink.
If EudraLink is used for sending an eCTD sequence, the entire sequence has to be zipped first. Some zip
formats are not widely readable and therefore a submission may be rejected if the zipped format cannot be
read by the agency. If in doubt, please check the intended format with the concerned NCA before sending.
Please also note there is a size limit, refer to the EudraLink User Guide for further details (request
). Applicants
should not split an eCTD sequence over more than one zip/EudraLink. Please note, in order to re-obtain the
correct eCTD structure, unpack or extract the zip-file and save the content on your local path system.
Otherwise the eCTD structure is not displayed in the correct way.
When using EudraLink, it is strongly recommended that the expiry date is set to the maximum (90 days) to
ensure that it can be opened during the process at the receiving authority. In addition, all information relating to
the submission must be contained within the zipped sequence; no formal information should be included in the
body of the EudraLink message.
2.10.6 Labelling/Metadata
The information provided on either the physical media or metadata provided when using a portal must be
consistent with the information provided in the cover letter and the eCTD envelope.
When using the CESP portal this metadata should go into the XML delivery file.
For submissions to EMA the relevant information is included in the eSubmission Gateway / Web Client
file
naming convention or XML delivery file or in the XML delivery file for PSUR submissions.
Each CD or DVD submitted should include the following label information clearly presented and printed on the
media:
Format: eCTD
The applicant’s name
The product (invented) name(s)
The International Non-proprietary Name (INN) of the active substance(s)
The full procedure number(s) (if known) (e.g. UK/H/1234/001-005/II/0034)
Technical eCTD Guidance v4.0 Page 18 of 62

The sequence number(s) of the eCTD submissions contained on the CD/DVD
If there are too many sequences to list on the CD/DVD label itself, a separate list should be
provided in the cover letter.
Number of media units per full set and an indication of the place of the individual CD/DVD within this
set (e.g. 1(5), 2(5), etc.).
The submission type(s) of each eCTD submission(s) contained on the CD/DVD (e.g. Initial Application,
Variation Type II), as per the eCTD envelope information
2.11 Number of Media Requested
The details of the number of copies for media required for archiving and reviewing purposes are published on
the CMDh website and under
question ‘How and to whom should I submit my dossier?’ of the Human/Pre-
authorisation Q&A on the EMA’s website. Where an NCA requires the disc to be archived they may have
additional requirements.
2.12 Technical Baseline Applications
A baseline submission is a compiled submission of the current status of the dossier, i.e. resubmission of
currently valid documents that have already been provided to an agency but in another format. The sections
provided to make up a baseline can be defined by the applicant, but any omissions should not render the
submitted content misleading. A baseline would typically consist of the Module 3 documents that tend to
change over time during the lifecycle of the product.
It is highly recommended but not mandatory to use a baseline as a start of an eCTD when changing from paper
or NeeS and to provide as much content as possible in the eCTD. The baseline would preferably consist of
high quality electronic source documents, but good quality scanned images would also be acceptable in these
cases, preferably with Optical Character Recognition (OCR) to facilitate text searching.
It should be clearly stated in the cover letter of the “baseline eCTD sequence” that the content of the previously
submitted dossier has not been changed, only the format. There is no need for the NCAs or EMA to assess
baseline submissions and hyperlinks between documents are not needed. The submission type reformat
should be used in the envelope for the baseline sequence.
2.12.1 Baselines Starting as Sequence 0000
The baseline should normally be submitted as sequence 0000, but could in some justified situations also be
submitted at a later stage (see Section 2.12.2 below). The baseline should always be a separate submission
and should never include new applications. The first new regulatory activity, e.g. the next variation, in eCTD
format should then be submitted as sequence 0001, see table below.
Table 5 : Example for starting an eCTD with a baseline sequence
Sequence
number
Submission Description
Submission Type
Related
Sequence
Submission
Unit
0000
Baseline of Module 3
none
0000
reformat
0001
Variation for new indication of
COPD
var-type2
0001
initial
0002
Response to Questions
var-type2
0001
response
0003
Variation to shelf life
var-type1b
0003
initial
0004
Extension for 8mg tablet
extension
0004
initial
2.12.2 Baselines Starting Later in Lifecycle
A baseline can also be submitted later in the lifecycle. If documents have already been provided in previous
submissions in the sections now covered by the baseline, these should not be re-submitted. Instead, the
remaining incomplete sections should be filled up with earlier dossier content (paper or NeeS), now provided in
eCTD format for the first time.
Technical eCTD Guidance v4.0 Page 19 of 62

It is possible to use multiple sequences to submit a baseline, e.g. one sequence for the baseline for Modules 4
and 5 followed later by one sequence for the baseline for Module 3. The submission type ‘reformat’ should be
used in each case. An example is given below.
Table 6 : Example for starting an eCTD with regulatory activity sequence
Sequence
number
Submission Description
Submission Type
Related
Sequence
Submission
Unit
0000
Variation concerning Modules 4
& 5
var-type2
0000
initial
0001
Variation for new indication of
COPD
var-type2
0001
initial
0002
Response to Questions
var-type2
0000
response
0003
Baseline of Module 3
none
0003
reformat
0004
Extension for 8mg tablet
extension
0004
initial
In cases where a product, nationally approved in more than one EU country, becomes an MRP product through
a referral, it is quite likely that the eCTD dossiers submitted nationally are incompatible and thus cannot be
used to continue the MRP dossier. The dossier might then have to start anew, from sequence 0000 and be
compiled in line with CMDh guidance ‘
Requirements on submissions for New Applications within MRP, DCP or
National procedures’. In such cases, a baseline submission might be justified in order to give all the CMSs
access to the previously submitted documentation. For details on how to transfer existing eCTD lifecycle from
one procedure to another (e.g. at the end of an Article 30), see Section 2.12.3 below.
For eCTD dossiers created with old tools and/or in accordance with technical criteria which are now outdated, a
baseline can be submitted in order to “clean up” the dossier from any technical issues that would cause
problems. However, the applicant should first ensure that there are no other ways of rectifying these technical
issues so that this option is not used unless absolutely necessary.
The technical baseline application can also be used by applicants to switch from one eCTD sequence per
strength to one eCTD sequence covering multiple strengths (see Section 2.12.3 below). For the switch, the
pros and cons of the different approaches to dossier structure, as described in Annex 3
, should be taken into
consideration. The switch from one approach to another should normally only be allowed once during the
lifecycle, and must be agreed by the relevant authority.
2.12.3 Re-Baselining a Broken eCTD Lifecycle
One of the principles of eCTD is that with the use of the operation attributes, it is possible to manage the
lifecycle of a product and generate a view of the “current dossier”.
However, in certain cases, the lifecycle at the side of the applicant may be broken.
This situation can occur in cases such as:
• An MA is transferred to another MAH who is unable to import any existing eCTD sequences into its
building tool
• An applicant switches to a new publishing tool and is unable to import their submitted sequences
• An applicant is working with a lifecycle where previously submitted sequences are actually invalid, and
were not tested at the time by the receiving agency
The problem with all of these situations is that the applicant cannot continue with the existing lifecycle of the
product. Any subsequent submission (sequence) for the product where previously submitted content is
changed and needs to be referred to (using the operation attributes replace, or delete) cannot be built in the
tool, or, if built, would be invalid. This is because it is impossible to create the link back to the original submitted
documentation, because it no longer resides in the eCTD building tool.
In these first three examples, the preferred situation would be that the previous submitted sequences are
imported in the new tool and the lifecycle of the product will continue. However, this might not be possible, due
Technical eCTD Guidance v4.0 Page 20 of 62

to technical issues in uploading previous sequences into a different tool, or particularly when the previous
sequences were invalid. If the lifecycle re-starts a new UUID for the application is required.
In addition, in exceptional cases, there may be a benefit to both the applicant and to the agency if the current
lifecycle is archived in some way and re-started. For example:
• An applicant has chosen in the past to submit more eCTD applications than needed under current
guidelines, for example, one for each strength of a product
• An applicant has used the parallel national model in MRP/DCP and needs to switch to the
comprehensive model
• At the end of an Article 30 procedure, the applicant is switching from national eCTD in one or more MS
to a comprehensive eCTD for the new MRP
In order to ensure that in future the lifecycle of the product is correctly maintained, it is proposed that in these
exceptional circumstances, and with prior agreement between the MA holder and the receiving NCA (national
procedures) the RMS (MRP/DCP), or EMA (centralised procedures), applicants are able to resubmit the current
registered dossier as a baseline consisting of all valid documents as seen in “current view”, leaving the existing
sequences in place, but essentially resubmitting the content in a new eCTD application. Also in these cases a
new UUID for the application is required.
In the cover letter the applicant provides details of why the lifecycle is broken, and that a new eCTD sequence
is being submitted in order to restart the lifecycle.
• A new UUID need to be assigned to the application.
• The submission type would be “none”
• The submission unit type would be “reformat”.
• The operation attributes of the leaves would be all “new”.
• The sequence number of the submission would normally be restarted at 0000 and not continued from
the previous lifecycle, since continuation of existing numbering could lead to complex lifecycle issues.
However, if there is a valid eCTD lifecycle available that can be re-used, this should be considered by the
applicant – for example, if moving from the historical parallel national model to the comprehensive model in an
MRP/DCP, if one of the national countries has a number of valid sequences, these could be provided to the
remaining countries and used as a basis for the comprehensive eCTD. In those cases the UUID must not
change.
Also, when compiling several eCTDs built per strength or form of a product into only one combined eCTD for
that product, normally one of the strengths lifecycle could be kept and be completed with the missing
documents from the current view of the other strengths to give the complete current dossier. In those cases the
assigned UUID of the application maintained will also serve for future lifecycle. Any re-use of existing
sequences or changes to sequence numbering should be agreed with the relevant authorities.
Re-baselining where the previously submitted sequences cannot be used in the new
lifecycle and must be archived
For the agency, the former submitted sequences have to be handled as “history”, and the new set of
sequences would need another identifier to be set by the authority to differentiate them from this previous
lifecycle. The lifecycle will begin from scratch again from the time of the baseline submission with a new UUID
in each sequence built with EU eCTD m1 v3.0 or later. In an MRP, there is no need to mention the previous
(archived) sequences in the tracking table, so the new tracking table should only refer to the re-established
lifecycle.
Scenario 1 – previously submitted sequences archived, new eCTD started
Applicant X has submitted sequences:
0000 Initial application
0001 Validation update
0002 Day xx response
0003 Day yy response
Technical eCTD Guidance v4.0 Page 21 of 62

0004 Variation 001
** Problem occurs in continuing lifecycle, see examples below
0005 0000 - Next submission
0005 is not submitted. Instead, 0000 - 0004 are archived, and a new eCTD is started at 0000.
Examples for this scenario:
** MA is transferred, previous sequences 0000-0004 cannot be imported into a tool by the new holder.
** Applicant changes their eCTD Building tool, previous sequences will not import into the new tool
** Previous sequences 0000-0004 were technically invalid according to the specification at the time, but were
accepted by the agency because eCTD checking was not yet established
** Sequences 0000-0004 were “not mutual” (parallel national) – not all countries in the procedure may have
received all of them with the same sequence number
Re-baselining where the previously submitted sequences in at least one MS can be
used as exisiting lifecycle
In the case that previous lifecycle can be continued, but submitted in additional member states, then there
would be no need to change the identifier or UUID. In an MRP, the tracking table should indicate which
member states originally had the sequences, and which member states are now getting them as lifecycle
history.
Scenario 2 – previously submitted sequences valid, lifecycle continued
Applicant X has submitted sequences:
0000 Initial application
0001 Validation update
0002 Day xx response
0003 Day yy response
0004 Variation 001
** Problem occurs in continuing lifecycle without making changes to the scope of the eCTD application,
examples below
0005 = Next submission
The original sequences are maintained, but a “new” eCTD lifecycle is started at 0005, where more countries
receive the lifecycle.
Examples for this scenario:
** Sequences 0000-0004 were “not mutual” (parallel national) – all countries in the procedure have received all
of the sequences as individual national sequences with the same sequence number
** Earlier sequences 0000-0004 referred to only one strength or dosage form, but the new lifecycle will cover
more multiple strengths/forms. Note there is no need to alter the metadata from the previously submitted
sequences, the additional strengths / dosage forms can be added in subsequent sequences.
** Earlier sequences 0000-0004 were used in national procedure prior to an Article 30 procedure, but can be
re-purposed for the new MRP
Technical eCTD Guidance v4.0 Page 22 of 62

3. MODULE SPECIFIC INFORMATION
3.1 General Information
The following subfolders should be used to organise the files for each module in a submission: m1, m2, m3,
m4, and m5 following the principles set out for the CTD in Notice to Applicants, Volume 2B
. There is also a
subfolder util to organise eCTD technical files in the submission. If a module is not appropriate for a particular
submission it should be omitted. Empty subfolders should not be included.
Each document should be provided as an individual PDF file, except those specifically requested in a different
format.
A single eCTD application can cover multiple drug substances (e.g. in case of fixed combination products),
multiple manufacturing sites, multiple medicinal products based on one invented name (different
pharmaceutical forms or strengths). Careful planning is required to ensure that the dossier can be expanded as
the product range is expanded or reduced by the submission of later sequences. Please see Annex 3
for
further details.
3.2 Module 1 eCTD Envelope, Administrative Information and Prescribing
Information Folder
3.2.1 General Considerations
In the case of country specific files or folders the country code should appear in the file and folder name as the
differentiating marking.
“Not Applicable” Module 1 documents should not be included in the eCTD. However, when a justification for
the absence of a certain document in Module 1 is required, such justification should be provided in its
corresponding section in the eCTD structure. In any case, all section titles should always appear in the
Module 1 eCTD backbone, displayed by the style sheet, even if these sections are not populated.
3.2.2 Creation and Management of Envelope Information
The eCTD envelope should be used to describe the eCTD sequence:
Country In the centralised procedure, there should only be one envelope, and this should
have the entry ‘ema’. For MRP/DCP, each country in the procedure needs to have a
separate envelope entry. ‘Common’ must not be used as a country identifier in the
envelope. In case of submissions to EDQM the envelope need display the entry
‘edqm’.
Identifier A UUID as specified by ISO/IEC 11578:1996 and ITU-T Rec X.667 | ISO/IEC 9834-
8:2005. The same UUID will be used for all sequences of an eCTD application.
Submission Type This value represents the regulatory activity which will be started and the type of
material sent to the agency. The entry is chosen from a picklist based on the
EUTCT controlled term list on Applicant Submission Type, see EU M1 Specification
for further details.
Submission Unit Submission unit type describes the content at a lower level (a “sub-activity”) which is
submitted in relation to a defined regulatory activity. The entry is chosen from a pick
list, see EU M1 Specification
for further details.
Submission Mode This element should only contain a value in variation, PSUSA or line extension
regulatory activities and must be included in every sequence of that activity. The
value can be set to ‘single’, ‘grouping’ or ‘worksharing’.
Submission Number This number represents the high level procedure number related to the regulatory
activity in case of worksharing submissions and for submissions of grouped Type
1A variations and PSUSA if multiple products are concerned. Samples are
provided in the EU M1 Specification
. Note: not required in case only one product is
concerned.
Technical eCTD Guidance v4.0 Page 23 of 62

If submission mode is set to either 'grouping' or 'worksharing' there should be a
Submission Number (Number element in the DTD). This Number element should be
identical with the variation procedure number included in a Variation eAF.
Procedure tracking
number Always to be completed. In case of using the submission number, the Procedure
Tracking Number (PTN) refers to the product involved in the worksharing, grouped
or PSUSA procedure. Any value used by an agency or applicant to track the
submission, in any procedure, in relation to a particular product, e.g.
EMEA/H/C/000123 for a CP submission (after allocation of the specified procedure
number by EMA) and DE/H/0126/001/MR for an MRP submission (depending from
the national business rules which might allocate procedure tracking numbers at
receiving time point only. In those cases the field remains empty until submission). If
the PTN is not known in advance, it is recommended to use the product number
instead. For a full list of expected number types per agency refer to EU M1
Specification. Note: When a submission is not relevant for all products covered by
the dossier or new products are added to the dossier, please clearly state this in the
cover letter.
This number should be congruent with the variation procedure number included in a
Variation eAF or with the procedure number included in a MAA eAF or in a Renewal
eAF.
Applicant Entries for ‘applicant’ should be consistent for all eCTDs from any single applicant
(legal entity), as they define where eCTDs are stored in internal systems.
Consistency of spelling is also relevant over time to allocate the eCTD correctly.
In case of ‘worksharing’ procedures, only the name of the applicant designated for
the worksharing submission should be used.
Agency Code Select from picklist in the most recent EU M1 Specification. Assure that Country and
Agency name will be consistent.
Procedure type The entry is chosen from a picklist, see EU M1 Specification
for further details.
Invented-name The trade name/invented name for the medicinal product covered by the
application. If the eCTD covers multiple strengths or dosage forms, this entry does
not need to describe the complete name, a simple entry, for example, ‘Wonderdrug’
will suffice.
INN The International non-Proprietary name for the drug substance.
Sequence The sequence number here must match the sequence number in the folder
structure, on the Cover letter and on the XML delivery file for CESP submissions,
the file naming conventions for eSubmission Gateway/Web Client submissions and
the XML delivery file for PSUR submissions via the eSubmission Gateway/Web
Client. If the submission is provided on CD/DVD this should match the label of the
CD/DVD.
Related-sequence For a description and example of how to use the ‘related sequence’ entry, see
Section 2.9.4 and the EU M1 Specification
.
Submission-description This element is used to describe this particular eCTD sequence.
3.2.3 Module 1.0 Containing Cover Letter and Tracking Table
3.2.3.1 Cover Letter
The cover letter should always be submitted with the document operation attribute “new”. As eCTD viewing
tools will display all "new" leaf elements in a current or cumulative view, it is recommended that additional
descriptive text is included in the leaf title to help identify specific cover letters. This will help identify each
cover letter leaf and the submission it is in, rather than having the cover letters named the same in each
sequence. Some examples for the leaf titles could be:
Cover Letter for Sequence 0000
Cover Letter for Germany for Sequence 0000
Cover Letter for France for Type II Variation 028 (0042)
Technical eCTD Guidance v4.0 Page 24 of 62

Please see also the CMDh website for requirements of signed paper copies of the cover letter and application
form to each NCA. Please note, when resubmitting content due to technical validation issues or sequences
missing at NCA side, a note (a comment in the delivery file (CESP, working documents or separate note in
case of CD/DVD submissions) may be provided to clarify the reason for resubmitting and the original cover
letter in the eCTD should not be changed, see
Section 2.9.4. For submissions to the EMA it is mandatory to
use the eSubmission Gateway / Web Client and no additional comment is required.
For CP there is
a cover letter template published on the EMA website under the Post Authorisation Guidance.
For MRP / DCP guidance is provided here.
3.2.3.2 Tracking Table
A tracking table should always be included as an annex to the cover letter for MRP and DCP. This is also
highly recommended for CP and NP. The file should be named cc-tracking-var.pdf and be placed in
/XXXX/m1/eu/10-cover/cc. (e.g. ema-tracking-var.pdf for the CP, common-tracking-var.pdf in an MRP/DCP, or
be-tracking-var.pdf in a NP.)
3.2.4 Application Forms
The application form should always be submitted with the document operation attribute “new” (as for the cover
letter, see above), unless an error has been made in the form and an updated application form is being
provided, in which case the operation attribute should be “replace”.
Some NCAs do require the application form to be submitted as a signed paper original together with the eCTD
submission. Some NCAs, on the other hand, request that applicants create a web-based application form on
their portals in addition to the electronic application form, which assist in their internal case creation process.
Please refer to the CMDh website
or the individual NCA’s web sites for further details on specific requirements.
The electronic Application Form (eAF)
, should be provided in PDF format only (with the extractable data),
inside the eCTD. The use of the eAFs is mandatory from 1
st
July 2015 for the CP applications, and from
1
st
January 2016 for all other procedures. These forms can be found from the electronic Application Forms
webpage.
The file named cc-form.pdf should contain the eAF in all cases (without using any variable part of the file
name).
Supportive documents, which are not part of any M2-5 section or Response to Questions, should be placed in
the application form section of the eCTD, and not appended to the form itself. Each annex should be provided
as a separate attachment in m1/eu/12-form/cc, using the variable part of the file name and the leaf title to
clearly describe the content of the document. E.g. file name: fr-form-proofpayment.pdf, Leaf Title: “France,
Proof of Payment“.
3.2.5 Product information
Product information should be supplied as PDF files within the eCTD. In addition Word files (with or without
tracked changes as relevant) should be provided in the separate folder XXXX-workingdocuments within the
same submission. Alternatively, certain procedures require word working files to be sent via Eudralink. Details
can be found in Section 2.9.9 and from Section 4. Advice on specific application types
.
Additional tracked changes version in PDF format are required only in the Centralised Procedure for either
product labelling or risk management plan documentation.
In MRP/DCP national translations should be managed outside of the eCTD (see CMDh guidance
‘Requirements on submissions for New Applications within MRP, DCP or National procedures
’).
In MRP/DCP, ensure that common product information is always placed under m1/eu/13-pi/131-
spclabelpl/common/en. If these documents are placed under country specific folders, e.g. the country folder of
the RMS, the agencies will not be able to use the country filtering options of their eCTD viewer tools as
intended.
Technical eCTD Guidance v4.0 Page 25 of 62

In the Centralised Procedure, national translations are only required in the eCTD for the commission decision
documents in a closing sequence. Please refer to Section 4.1
.
3.2.6 Use of Response Documents Section
The submission of electronic information in response to a list of questions from NCAs and EMA should follow
the same basic principles as the first submission. The written response should be submitted following the EU
recommended response folder and file structure. Please note that all data related documents are aligned with
the CTD structure; refer to EU NtA Presentation
and format of the dossier (CTD) Document, using the
operation attributes of “new”, “replace”, or “delete” as appropriate. (“Append” should be avoided, see Section
2.9.6.)
To help in the management of responses over the lifecycle of the eCTD, the responses relating to a particular
regulatory activity should be grouped under a node-extension in the eu-regional.xml file. The title of the node-
extension should identify the regulatory activity (e.g. Responses to Questions for the Initial Application,
Responses to Questions for Type II Variation 028, etc.). It is recommended that the applicant provides a full
copy of the list of questions received from the agencies as the first leaf in this section.
It is recommended that the responses be split up into separate files for each major section of the submission
(e.g. Quality, Non-clinical and Clinical). Leaf titles should be used to identify the particular set of responses
(e.g. Response to Major Objections - Quality). If responses to more than one question are submitted in a single
file bookmarks should be used within the PDF file to clearly identify each response. It is possible to submit the
response to each question in a separate file but in that case node-extensions must be used and leaf titles to
group and identify the responses under the top level node-extension.
In MRP/DCP, all of the files for the response documents should be placed in the folder
m1/eu/responses/common, regardless which member state raised the question.
In m1-responses/cc, it is recommended to use the variable component of the filename and the leaf title, to
present the information clearly to the assessor. The response files should preferably be named as:
cc-responses-<regulatory activity type identifier>-<timeline identifier>-<content identifier>.pdf, using the -var
component of the filename to define the content.
Table 7 : Examples of Filenames and Leaf Titles for Response Documents
Suggested Filename
Suggested Leaf Title
common-responses-maa-d106-clin.pdf
Day 106 Clinical Responses, MAA
common-responses-maa-d120-clin.pdf
Day 120 Clinical Responses, MAA
common-responses-maa-d145-clin.pdf
Day 145 Clinical Responses, MAA
common-responses-maa-d120-clinq4.pdf
Day 120, Clinical Response Question 4, MAA
common-responses-maa-d120-clinq7.pdf
Day 120, Clinical Response Question 7, MAA
common-responses-var03-d45-clin.pdf
Day 45 Clinical Responses, Var03
common-responses-var03-d59-clin.pdf
Day 59 Clinical Responses, Var03
common-responses-maa-d106-qual.pdf
Day 106 Quality Responses, MAA
common-responses-maa-d120-qual.pdf
Day 120 Quality Responses, MAA
common-responses-maa-d145-qual.pdf
Day 145 Quality Responses, MAA
common-responses-var05-d44-qual.pdf
Day 44 Quality Responses, Var 05
common-responses-var05-d59-qual.pdf
Day 59 Quality Responses, Var 05
common-responses-var12-d33-qual.pdf
Day 33 Quality Responses, Var 12
Technical eCTD Guidance v4.0 Page 26 of 62

3.2.7 Use of the Additional Data Section
The section 'Additional Data’ should only be used for nationally required information in National, Mutual
Recognition and Decentralised Procedures. An exemption to this is the use of this section for justifications for
active substances or justification of eligibility of the product for the Centralised Procedure.
In addition this section can be used for all procedures when an old version of a DTD is being used during an
agreed transition period, to support inclusion of a newly defined section of Notice to Applicants (refer to
transition guidance issued with specification updates).
3.3 Module 2 Overviews and Summaries Folder
3.3.1 General Considerations
Each document should be provided as an individual PDF.
3.3.2 Structure of Module 2 Documents
In module 2.3 Quality Overall Summary either one file (qos-var.pdf) or separate files per QOS section can be
submitted named as: introduction-var.pdf, drug-substance-var.pdf, drug-product-var.pdf, appendices-var.pdf
and regional-information-var.pdf. For details refer to the ‘File-Folder Structure & Names’ tab in the EU
Validation criteria spread sheet.
If an existing document is revised, the lifecycle operator ‘replace’ should be used. However, if changes of
content only affect one section of that document then updates should normally be submitted as a separate
summary with the document operation attribute “new” as it would help clarifying what to assess within the
specific submission.
For submissions covering multiple indications, refer to Section 3.6.1
.
3.4 Module 3 Quality Folder
3.4.1 Module 32S drug substance
If the product contains multiple drug substances, then documentation for each substance should be provided in
its own m32s section. If a drug substance is manufactured at multiple sites or by multiple different
manufacturing companies, documentation can be provided in multiple m32s sections. However, it may be
possible to write documentation that covers multiple manufacturers in one CTD section – the way the
information is provided is left up to the applicant. For further details, please see Annex 3
.
3.4.2 Module 32p drug product
Each dosage form covered by an eCTD application should be described in its own m32p section. If an
application describes multiple strengths of any one dosage form, then documentation that covers all strengths
can be provided in a single m32p section. Alternatively, each strength can be covered by its own strength-
specific documents in multiple strength-specific CTD sections. For further details, see Annex 3
.
3.5 Module 4 Nonclinical Study Reports Folder
3.5.1 Guidance on the Handling of Granular Study Reports
Submissions created in eCTD format for the use within the FDA may provide more granular study reports using
study tagging files. There is no need to re-organise the reports for submission to the EMA or NCAs. See
Section 3.6.2.
below for further information.
Technical eCTD Guidance v4.0 Page 27 of 62

3.6 Module 5 Clinical Study Reports Folder
3.6.1 Management and Handling of Multiple Indications
In cases where the application includes multiple therapeutic indications, the reports should be organized in a
separate Section m535 for each indication. In such cases, if a clinical efficacy study is relevant to only one of
the indications included in the application, it should be included in the appropriate section in m5 (e.g. m5/53-
clin-stud-rep/535-rep-effic-safety-stud/anxiety/5351-stud-rep-contr). If a clinical efficacy study is relevant to
multiple indications, the study report should be included in the most appropriate subsection of m535 and
referenced as necessary in the equivalent section under the different indication. In Module 2, a separate
“Summary of Clinical Efficacy” module should be submitted for each indication, although closely related
indications can be within a single document.
Regardless of which way is chosen, it is important to provide clear written guidance to the assessor when the
supportive data/study report documents are applicable to more than one indication.
3.6.2 Management and Handling of Granular Clinical Study Reports
ICH Q&A 22 recommends use of E3 granularity for clinical study reports. In Europe, node extensions should be
used to group together individual files. STFs from submissions in the US are not required but a submission will
not be rejected if they are included. If a US NDA is repurposed for submission in the EU, the study content (the
study report and any relevant appendices) should be placed under a node extension. Ideally, the STF xml file
itself and any content not usually provided in Europe (e.g. datasets) should be removed. In order to maintain a
consistent looking eCTD lifecycle and table of contents (via index.xml), applicants are advised to use node
extensions for all clinical study reports, regardless of the granularity of the content (i.e. even reports that consist
of only one document should also be presented in node extensions). See also Section 2.9.8
Node-extensions.
3.6.3 Provision of CRFs and Data when Requested
If case report forms and individual patient data listings are submitted in m537 (as appendices 16.3 and 16.4 in
the ICH clinical study report guideline E3) they should be placed in the same order as the clinical study reports
appearing in m535 and should be indexed by study. Please note that bookmarks will not be required as there
will be no further internal structure.
3.6.4 Provision of Synopses of Individual Studies
It is acceptable either to include copies of the synopses for each study in eCTD section 2.7.6 or to provide
hyperlinks to synopses located in Module 5 without providing copies in section 2.7.6. In either case a Listing of
Clinical Studies should be provided and this should include hyperlinks to the first page of each synopsis.
3.6.5 Company Core Data Sheet
If companies submit their Company Core Data Sheet, this is recommended to be provided in eCTD section
5.3.6, Post Marketing Experience.
Technical eCTD Guidance v4.0 Page 28 of 62

4. ADVICE ON SPECIFIC APPLICATION TYPES
4.1 New MA Applications
The recommended start for an eCTD lifecycle is the initial MA application. It should normally be provided as
sequence 0000. To start with another number should be justified in the cover letter. All documents included
should have the operation attribute “new” and be placed in the relevant sections in line with the different eCTD
specifications.
The submission type should be ‘maa’.
See Section 2.9.5
for an example on the use of the submission units.
For responses to questions documents, see Section 3.2.6
.
The following milestones of the procedures are proposed as appropriate sequences to be submitted during the
assessment of a new application.
Table 8 : MAA – Centralised Procedure
Day Number/
Milestone
eCTD milestone sequence
Notes
Submission
deadline
Initial submission
As per published submission calendars
-5 or as
requested
before date of
start
Response to business validation issues
If required
121
Response to List of Questions (LoQ)
If applicable
181
Response to List of Outstanding Issues
(LoOI)
If applicable
Any time
between
D181-220
Redaction Proposal Document Package
For details see Section 4.12
Commission
Decision + 15
days or prior
to the next
regulatory
activity
whichever is
first.
Decision / Closing sequence – including
final translations
Updates to the dossier which have not yet
been submitted in eCTD but which have
been agreed by the CHMP at the time of
the opinion; e.g.
• Final RMP
• Minor updates to Module 2 or 3
• Final Product Information (Annex I,
II, IIIA, IIIB and Annex A) in all
languages
• Final mock-ups reviewed during
the procedure.
I.e. final amended documentation if any
changes occur during the Standing
Committee phase (SCP)
Except from changes during the SCP,
the documentation submitted within this
eCTD sequence should be identical to
the documents submitted to the EMA at
the time of the CHMP opinion via
EudraLink.
20 calendar
days post
consultation
notification
Final Redacted Document Package
For details see Section 4.12
Technical eCTD Guidance v4.0 Page 29 of 62

Table 9 : Centralised Procedure - Outside eCTD via EudraLink
211 (opinion
+ 1)
Final English PI
Opinion + 5
Provision of translations
Opinion + 25
Provision of final agreed translations
following linguistic review
Table 10 : New MAA – Decentralised Procedure
Day Number/
Milestone
eCTD milestone sequence
Notes
- 10
New MAA
Procedure
start
Validation update
If required
106
Day 106 Responses to questions
210
Final agreed EN product information
Or at any day when the procedure can
be closed after agreement is reached.
For further details on MRP and DCP, please refer to the specific CMDh guidance ‘
Requirements on
submissions for New Applications within MRP, DCP or National procedures’.
4.2 Variation Applications
All types of variations should be submitted within the eCTD as new sequences.
Documents related to the variation should be included in relevant sections or be deleted or replaced by use of
the appropriate document operation attribute. Where documents cannot be assigned to specific CTD defined
locations, then they should be attached to the 1.2 Application Form.
The submission type should reflect the type of variation. (See Q&A for Variations in eCTD
). Submissions for
workshare/grouping variations concerning several eCTD submissions are recommended to be supplied
together on a single CD/DVD. The CD/DVD should contain clearly marked subfolders for each product that
takes part in a worksharing or grouping procedure.
See Section 2.9.5
for an example on the use of the submission units.
For details on how to handle parallel variations, please refer to Annex 4
of this guidance.
Although Type IA variations usually should not be revised if they are invalid from regulatory point of view, it is
necessary to correct technical invalid submissions in the same way as required for any other eCTD sequence.
The following milestones of the procedures are proposed as appropriate sequences to be submitted during the
assessment of variations. Although the example relates to the Centralised Procedure the principal could be
applied to other procedures (except for final translations).
Table 11 : Type II Variations – Centralised Procedure
Day Number /
Milestone
eCTD milestone sequence
Notes
Submission
deadline
Initial submission
Please observe the published
submission timetables, e.g. “Start of
the procedure, new indication”
Technical eCTD Guidance v4.0 Page 30 of 62

-5 or as
requested
before date of
start
Response to business validation issues
If required
RSI
Submission
deadlines
Response to Request for Supplementary
Information (RSI)
If applicable
Opinion + 30
For Type II variations which are not followed
by an immediate Commission Decision:
Decision / Closing sequence – including final
translations if applicable
Updates to the dossier which have not yet
been submitted in eCTD but which have
been agreed by the CHMP at the time of the
opinion; e.g.
• Final RMP
• Minor updates to Module 2 or 3
Final Product Information (Annex I, II, IIIA IIIB
and Annex A) in all languages
For Type II variations which are not
followed by an immediate
Commission Decision
Commission
Decision + 15
days or prior
to the next
regulatory
activity
whichever is
first.
For Type II variations following worksharing
or with immediate Commission Decision:
Decision / Closing sequence – including final
translations if applicable
Updates to the dossier which have not yet
been submitted in eCTD but which have
been agreed by the CHMP at the time of the
opinion; e.g.
• Final RMP
• Minor updates to Module 2 or 3
• Final Product Information (Annex I, II,
IIIA IIIB and Annex A) in all
languages
•
Final mock-ups reviewed during the
procedure.
For Type II variations following
worksharing or with immediate
Commission Decision
Table 12 : Type IA & IB Variations – Centralised Procedure
Day Number
eCTD milestone sequence
Notes
Submission
Start of the procedure <description>
e.g. “Start of the procedure, phone
number changes”
RSI
Submission
deadlines
Response to Request for Supplementary
Information (RSI)
If applicable
Table 13 : Type IB Variations with linguistic review - Centralised Procedure
Day Number
eCTD milestone sequence
Notes
Submission
deadline
Start of the procedure <description>
English PI (pdf) – inside the eCTD
English PI + translations (word) (Outside
eCTD but within the same submission
package in the workingdocuments folder)
e.g. “Start of the procedure, phone
number changes”
Technical eCTD Guidance v4.0 Page 31 of 62

Start + 5
Provision of translations (Eudralink)
Start + 25
Provision of final agreed translations
following linguistic review (Eudralink)
RSI
Submission
deadlines
Response to Request for Supplementary
Information (RSI)
If applicable
Notification +
15
Closing sequence – including final
translations (pdf) inside the eCTD
For MRP and DCP, please refer to the CMDh guidance ‘
Requirements on submissions for New Applications
within MRP, DCP or National procedures’.
4.3 Extension Submissions
Several dosage forms can be managed within a single eCTD application, and this helps to avoid submission of
data multiple times (e.g. active substance changes). Submissions for an extension can either be submitted
within an existing eCTD application, as a new sequence (continuous sequence numbering), or as a new eCTD
application (sequence 0000), if a separate lifecycle management is preferred (not applicable in the Centralised
Procedure, see below).
In MRP/DCP, an extension will be submitted within the same procedure, but with a different product number,
and as such, the recommendation is to submit the extension as a new sequence within the original eCTD
application, submitting a new Module 1, an updated Module 2 and new or updated 32P section. If m32p is
combined for all previous existing strengths/dosage form(s), an updated section should be provided, replacing
existing documents where necessary. If a separate m32p is being provided for the additional strength/dosage
form to describe the extension, then all documents should have the operation attribute of 'new'.
For extension applications, only new data should be submitted as a new sequence in the already submitted
eCTD. The submission type should be “extension”.
If single eCTDs are used for each strength or form of a product, full data concerning the extension applied for
has to be included in the submitted eCTD and therefore clear information should be given to the assessor on
what is new compared to earlier submitted data for the product to avoid unnecessary assessment.
In the Centralised Procedure, extensions are typically managed under the same procedure number as the
original dosage form, and again the recommendation is to submit the extension as a new sequence within the
original eCTD application, using a new m32p to describe the different dosage form.
For national applications, the applicant should discuss with the relevant NCA.
4.4 Renewal Submissions
Please note that a renewal application can be used as the first eCTD in a product lifecycle in a similar manner
to variations. The recommendation given in the section above applies likewise.
The submission type should be “renewal”.
See Section 2.9.5
on examples on the use of the submission units.
The following milestones of the procedures are proposed as appropriate sequences to be submitted during the
assessment of renewals: Although the example relates to the Centralised Procedure the principal could be
applied to other procedures (except for final translations).
Technical eCTD Guidance v4.0 Page 32 of 62

Table 14 : Renewals
5 year Renewal – Centralised Procedure
Day Number
eCTD milestone sequence
Notes
Submission
deadline
Initial submission
As per published submission calendar
91
Response to Request for Supplementary
Information (RSI)
If applicable
Commission
Decision + 15
Decision / Closing sequence – including
final translations if applicable
Updates to the dossier which have not yet
been submitted in eCTD but which have
been agreed by the CHMP at the time of
the opinion; e.g.
• Final RMP
• Minor updates to Module 2 or 3
• Final Product Information (Annex I,
II, IIIA, IIIB and Annex A) in all
languages
Annual Renewal of conditional marketing authorisation – Centralised Procedure
Day Number
eCTD milestone sequence
Notes
Submission
deadline
Initial submission
As per published submission calendar
Table 15 : Centralised Procedure – Outside eCTD via EudraLink
Opinion + 1
Final English PI
Opinion + 5
Provision of translations
Opinion + 25
Provision of final agreed translations
following linguistic review
For renewals in MRP and DCP, the general principles in the CMDh guidance ‘Requirements on submissions for
New Applications within MRP, DCP or National procedures’ can be applied.
4.5 PSURs
As per the Article 107b paragraph 1 and Article 28(2) Regulation 726/2004) all Periodic Safety Update Reports
(PSUR) shall be submitted electronically.
The PSUR is a part of the product lifecycle and for products with eCTD lifecycle the PSUR must be submitted
in eCTD. The PSUR should be provided as the next relevant sequence in the products lifecycle. This applies to
all products, also for those for which the PSUR is part of an EU Worksharing procedure or the EU PSUR Single
Assessment (PSUSA) procedure.
Centrally Authorised Products (CAPs) for which the PSURs are submitted as part of the PSUR single
assessment should be submitted in eCTD format into their existing eCTD lifecycle.
Nationally Authorised Products (incl. MRP/DCP/NP) that are submitted to the EMA as part of the PSUR single
assessment need to be submitted in eCTD or NeeS format as appropriate.
A separate eCTD sequence must be submitted for each respective product/presentation with its own lifecycle.
Technical eCTD Guidance v4.0 Page 33 of 62

If a single PSUR has been prepared covering multiple products for which the lifecycle is in eCTD format, a
separate submission (of that same PSUR document) must be made for each respective product. Separate
standalone eCTD sequences grouping multiple products should not be created.
The submission of a PSUR should consist of a cover letter and the report itself as a new document in m536 as
well as a new or replace document in m25 as necessary. Any literature will be included in m533 or m54 as
appropriate. The naming of the leaf element in m536 shall indicate the number of the PSUR or the period
covered. In case of proposed changes to the product information texts, new (if there is no previous lifecycle in
the product information section) or replace versions need to be submitted in m131 in a closing sequence. The
closing sequence usually contains all the agreed translations in all languages.
When multiple products from the same MAH are submitted, all products must be listed in the cover letter. It
should also be clarified in the cover letter that the content of each sequence/submission is identical. For PSUR
Worksharing procedures, the same principles apply. However, exemptions to this principle might be necessary
in the future, and if so please refer to any specific guidance from EMA and NCAs.
More information on the use of the cover letters for the PSUR submissions can be found from the EMA
Post-
Authorisation Guidance.
The submission type should be “psur” for ‘pure, single PSURs’, i.e. those products for which the active
substance is not included in the EURD list.
For PSURs included in the EU PSUR Single Assesment (PSUSA), the submission type is “psusa”
See Section 2.9.5
for an example on the use of submission unit.
Please refer to the PSUR Repository website
.
Note: MAHs should not submit full study reports as part of a PSUR. Final study reports should always be
submitted as a C.1.13 variation. In the case of interim PASS study results, these can be summarised in the
PSUR under the section “Findings from non-interventional studies” or alternatively if the reporting to EMA does
not coincide with the PSUR submission, the full interim report should be submitted as a separate, stand-alone
submission (post-authorisation measure (PAM)) relevant to CAPs only.
Additionally, submission of RMP updates cannot be accepted with PSURs subject to a PSUSA of:
• a mixture of CAPs pertaining to different GMAs;
• a mixture of centrally and nationally authorised medicinal products;
• a mixture of NAPs.
The submission of an imposed, non-interventional Post Authorisation Study protocol or study report should not
be combined with a PSUR or PSUSA sequence. Please, use the submission type ‘pass107n’ in case of the
protocol and ‘pass107q’ in case of the study report.
4.6 Referrals
4.6.1 Referrals handled through CMDh:
The response that the applicant has to prepare to the list of questions prepared by the CMD(h) will be sent as
an eCTD sequence to all CMD(h) members, according the timelines as defined. The applicant will create this
new sequence in which the documentation is stored according to the recommended CTD format.
The type of submission of the new sequence should be “referral”.
4.6.2 Referral procedures for Centrally Authorised Products (CAPs):
eCTD format is mandatory for all submissions for CAPs involved in the referral procedure. Submissions should
be sent via EMA eSubmission Gateway or eSubmission Web Client only. All eCTD format submissions for
CAPs sent to EMA via eSubmission Gateway/Web Client are available via the Common Repository and will be
considered delivered to all National Competent Authorities’ (NCAs) representatives, alternates and scientific
Technical eCTD Guidance v4.0 Page 34 of 62

experts. No additional copies of the submissions should be sent directly to the NCAs on CD/DVD or via CESP
as this might lead to validation issues and cause delays.
CAP referral submissions should always be submitted as the next sequence in the product lifecycle for each
CAP. Standalone eCTD submissions for the active substance are not allowed for CAPs included in referral
procedures.
If the applicant submits new documentation/information, a new eCTD sequence should be created and
submitted. The applicant should not submit the entire history of all sequences (unless requested by the
authorities), but a new sequence with only the information/documentation that concerns the referral.
The type of submission of the new sequence should be “referral”.
See Section 2.9.5, tables 1 to 6 on examples on the use of the submission units.
In case of a newly created/submitted sequence, the cover letter contains background information for the reason
of the referral. Any other document, which concerns the referral, has to be included according to the CTD
structure (please refer to CTD structure in 4.6.3). Any additional documentation that doesn’t have a place in the
dossier, for example overview of the registrations/applications involved in the referral, should be placed in m10-
cover.
4.6.3 Referral procedures for Nationally Authorised Products (NAPs):
All submissions for NAPs involved in the referral procedure should be sent to EMA via eSubmission Gateway
or eSubmission Web Client. EMA strongly recommends sending all NAP submissions in eCTD or NeeS format.
Submissions for NAPs, sent in any format, are not available via the Common Repository and should be sent
separately to each NCA via Portal or on DVD/CD-ROM as applicable (please refer to
Dossier requirements for
referral procedures).
More information on the referral submissions can be found on the eSubmission Gateway webpage
. For more
information and updates please refer to eSubmission website.
There is no need to send any separate paper cover letters for these submissions, as the cover letter will be in
the relevant part of eCTD/CTD module 1 in PDF format.
4.7 Active Substance Master Files
For MAAs in eCTD format, applicants should incorporate the applicant’s part (AP) documents of the ASMF into
the eCTD structure as per the relevant guidelines. This is applicable even if the ASMF itself is not handled in
eCTD format. It is recommended to use a suffix of ‘-ap’ on the filename of these documents.
For further information, please refer to the specific eASMF page
on the eSubmission website.
A copy of the "Letter of Access" addressed to the regulatory authority included as Annex 6.10 of the application
form should be placed in m12/cc (i.e. in the respective folder for each concerned NCA).
4.8 Vaccine Antigen Master Files
The VAMF consists of the scientific data according to Part III of Annex I of Commission Directive 2001/83/EC
as amended. To support the lifecycle, keep the documents manageable and to assure the correct alignment of
the complete VAMF it is required that the manufacturer submits the VAMF (including versioning), preferably in
an electronic format following the principles of eCTD. The complete VAMF can be processed with its own
submission / case / procedure number separately.
The application of a medicinal product will contain the same data package including the certificate of
compliance with Community legislation, together with the evaluation report attached.
Technical eCTD Guidance v4.0 Page 35 of 62

4.9 Plasma Master Files (PMFs) and medicinal products containing PMFs
4.9.1 Plasma Master Files
The Plasma Master File documentation for certification is submitted to the EMA as a stand-alone eCTD dossier
of a medicinal product. This documentation contains all the information related to the starting material for the
manufacture of the medicinal product but submitted separately from MAA dossier (EC Regulation Commission
Decision 2003/83/EC, part III of Annex I) when follows the PMF certification evaluation procedure.
ePMF should be provided within the ‘workingdocuments’ folder for PMF certification submissions.
For practical on PMF eCTD guidance, the reader is referred to the see the PMF eCTD Guidance document
found via this link. More information on PMF submission can be found here
.
4.9.2 Medicinal products containing PMFs
All concerned medicinal products that include one or more plasma supplier in their dossier (i.e. one or more
PMF(s)) are required to have their dossier updated and implement this update submitting the relevant
variations to the MA (for eCTD guidance, see point 4.2 of this guideline).
4.10 Applicant Initiated Withdrawals of the MA or certain strengths, dosage forms
Applicants may decide to withdraw their marketing authorisation during any stage of the product lifecycle and
this section explains the general principles that should be followed in terms of managing the eCTD lifecycle.
If the application for withdrawal does not include all strengths and/or dosage forms covered by the same eCTD,
the application should be submitted as a new sequence, with the operation attribute “delete” for documents that
are no longer relevant. Furthermore, if relevant, it should also include updated documents with the operation
attribute “replace” for documents that covered several other strengths and/or dosage forms and that now need
to be revised to exclude the withdrawn strengths and/or dosage forms from the document.
Withdrawal of the whole product (all dosage forms and strengths) covered by an eCTD should preferably be
submitted as a new sequence only including a cover letter. The operation attribute “delete” is not required to be
used for the documents.
The submission type ‘withdrawal’ or the relevant variation category for the change, depending on the regulatory
procedure being followed should be used.
4.11 Applicant Withdrawal or Agency Rejections of Post-Authorisation Regulatory
Activities
If a regulatory activity is withdrawn, fully or partially or rejected fully or partially, then the documentation has to
be updated accordingly with a consolidation sequence. Documents that are no longer relevant should be
deleted, and documents where the content needs to be adjusted to reflect the withdrawal or rejection replaced.
A consolidation sequence should be submitted to restore the current view of the dossier to what it was before
the rejected activity was submitted.. This would also apply where documentation needs to be adjusted as a
result of a commitment or a partially rejected variation. The submission type should be ‘consolidating’.
Note, it is not possible to delete a sequence from the life cycle to accommodate the withdrawal or rejection.
Scenario on consolidation sequence
Applicant X has submitted sequences:
0027 Variation 011
0028 Day xx response
Letter of rejection of variation 011
**0029 consolidating sequence to restore the previous status of dossier
0030 Variation 012
0031 = Next submission
Technical eCTD Guidance v4.0 Page 36 of 62

The variation 011 has been rejected and those parts affected by the proposed variation need to be restored to
present the previous status in the current view of the eCTD.
Examples for this scenario:
** After full or partial rejection of sequence 0027, (variation 011), a following sequence 0031 may change the
same technical content. If the change previously applied for in variation 011 has not been fully restored, the
new variation may not be displayed correctly, for example, the next submission cannot refer to content which
was proposed in variation 011 but subsequently removed. Therefore, the consolidating sequence, 0029, must
remove any new and now rejected content from the rejected variation, and also reinstate any documents that
were deleted or replaced in sequence 0027/0028, such that sequence 0030 can itself replace or delete the
content as required. i.e. Sequence 0029 must restore the current view, (in terms of what is current, not deleted
or replaced), to what it was before sequences 0027 and 0028 were submitted.
4.12 Publication of Clinical Data for Medicinal Products
Clinical reports will be published, under Policy 0070 (The European Medicines Agency policy on the publication
of clinical data for medicinal products for human use) following conclusion of the regulatory decision-making in
the frame of centralised marketing authorisation procedures.
Key components of the process from the point of submission by an applicant/MAH to the point of publication
are following:
“Redaction Proposal Document” package (eCTD sequence to be included in the eCTD lifecycle with the
attribute « new »)
“Final Redacted Document” package (eCTD sequence to be included in the eCTD lifecycle with the attribute
« new »)
For further details please refer to the Guidance for the publication of clinical data
:
4.13 Duplicate Applications and Informed Consent Applications
One eCTD per so called duplicate application has to be submitted and maintained separately in the post-
authorisation lifecycle phase (with the possibility of including several strengths, pharmaceutical forms etc. if
relevant). It should however be clearly written in the cover letter that it is exactly the same content (with the only
exemption of different tradename and maybe different MAH), so that redundant review work is avoided.
Duplicates are independent marketing authorisations and therefore the eCTD lifecycle will need its own UUID.
The term is used for practical reasons and understood as a MA application which is a ‘copy’ of another
application. Duplicates can be submitted under the same legal basis as the initial application (e.g. Art. 8(3) of
Dir 2001/83/EC). The legal basis of the dossier will trigger the dossier requirements. There is no definition of a
“duplicate” in the pharmaceutical legislation. However, for practical purposes, a duplicate application is defined
for MRP/DCP by reference to the first application or marketing authorisation as follows based on
CMD(h)
recommendations on multiple / duplicate applications:
– same dossier (copy of modules 1, 2, 3, 4 and 5);
– same legal basis according to Directive 2001/83/EC, as amended;
– different trade name;
– same or different applicant/marketing authorisation holder.
For CP specific requirements on multiple applications see the document ‘
Handling of Duplicate Marketing
Authorisation Applications’.
However, the life cycle for those dossiers needs to be handled as independent stand-alone dossiers with their
own life cycles and is following eCTD life cycle maintenance rules.
Applications under the legal base of Art. 10c – Informed consent will follow the same rules.
Technical eCTD Guidance v4.0 Page 37 of 62

Annex 1: eCTD Reference Documents
A number of relevant documents can be found on the Documentation tab on the eSubmission website at the
EMA. It is recommended that owing to the speed that information changes the following websites should be
consulted regularly in addition:
http://www.ich.org/products/electronic-standards.html
http://ec.europa.eu/health/documents/eudralex/index_en.htm
http://www.hma.eu/277.html
EU Module1 Specification
Most important documents to be considered are the following (as of March 2016):
• http://estri.ich.org/eCTD/eCTD_Specification_v3_2_2.pdf
• http://estri.ich.org/eCTD/eCTDQAV1_27.zip
• http://esubmission.ema.europa.eu/eumodule1/index.htm
EMA Pre submission guidance Q&As
• http://www.ema.europa.eu/ema/index.jsp?curl=pages/regulation/general/general_content_000157.jsp&
murl=menus/regulations/regulations.jsp&mid=WC0b01ac058002251f
EMA Post-authorisation Q&As
• http://www.ema.europa.eu/ema/index.jsp?curl=pages/regulation/general/general_content_000166.jsp&
mid=WC0b01ac0580023399
ICH M4
• http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/CTD/M4_R3_Organisation/M4_R3__orga
nisation.pdf
ICH M4 Q&As:
• http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/CTD/M4_R3_Organisation/M4_QAs.pdf
EU CTD Q&As:
• http://ec.europa.eu/health/files/eudralex/vol-2/b/ctd-qa-updatev3_2008-02_en.pdf
EMA Gateway:
• http://esubmission.ema.europa.eu/esubmission.html
Common European Submission Platform (CESP)
• http://cespportal.hma.eu
Electronic Application Form (eAF)
• http://esubmission.ema.europa.eu/eaf/index.html
Technical eCTD Guidance v4.0 Page 38 of 62

Annex 2: Guidance on Text Searchable Documents
A2-1 General
Applicants are requested to ensure that all submissions contain the maximum amount of text searchable
content. Documents with searchable text will aid the assessor, or any other user, in searching for specific terms
and also in copying and pasting information into another document, such as an assessment report.
It is recognized that not all documents need to be text searchable. This short document provides some
guidance about what must be text searchable and the ways to ensure that files are created appropriately.
A2-1.1 Creating Text Searchable Files
PDF files with searchable text can be created by all PDF tools from a source file in a text format (e.g. MS Word,
SAS, MS PowerPoint, Rich Text Files, etc.). When created in this way, the file will usually be the smallest in
size (measured in kilobytes or megabytes) that they can be.
If the only version of a document available is in paper, then scanning to PDF and using an Optical Character
Recognition (OCR) routine is the only way to create searchable text. PDF files created in this way tend to be
much larger in size, for the same number of pages (from 10 to 100 times as large), and the quality of the text
that is created will almost certainly not be a 100% match to the original text. It is noted that tools for checking
and correcting this text tend to be somewhat cumbersome. For these reasons, applicants are recommended to
use scanning/OCR only as a last resort.
Applicants are reminded that the text produced by the OCR routine should be “hidden” behind the image of the
original page so that the user can refer to the picture of the page and the text on it as final verification of the
data. As a result, the applicant should ensure that, as a minimum, the text on the scanned image is legible to
the user. Poor quality images should not be provided and you should note that these can only inevitably lead to
poor quality OCR text.
A2-2 Documents that Must Always Be Text Searchable
(i.e. the PDF should be produced wherever possible from a text source, such as MS Word, but if sourced from
a scanned original then they must be OCR’d.)
• Key administrative documents in Module 1 including, the cover letter, application form, product information
documents
o Applicants are reminded that some NCAs regard logging in through a portal as sufficient to
establish a user’s identity and do not require handwritten signatures on Cover Letters and/or
Application Forms submitted this way.
• Any document in Module 2 (QOS, Preclinical Overview and Summaries, Clinical Overview and
Summaries).
• The main body of text and main tables in any preclinical or clinical report required to support the main claim
of the application.
• The main body of text in any reports, methods, analytical procedures, etc. supplied in Module 3 The main
body of text of Periodic Safety Update Reports (PSURs)
• The main body of text of Risk Management Plans
• The main body of text of Environmental Risk Assessment
• Any English translation of a document originally written in a foreign language (see also below)
Technical eCTD Guidance v4.0 Page 39 of 62

A2-3 Documents that Do Not Need to Be Text Searchable
(i.e. the PDF should be produced wherever possible from a text source, such as MS Word, but if sourced from
a scanned original then there is no need for OCR.)
• Any original GMP certificate
• Any original certificate of analysis
• Any manufacturer’s licences
• Any certificate of suitability
• Any Manufacturing Authorisation
• Any document written in a foreign language where a translation is provided in English (however, the
translation should be text searchable, see above)
• Any literature references sourced from journals, periodicals and books (except when these are used in a
bibliographic application to support the main claims of the application).
• The blank CRF in a Clinical Study Report
• Patient data listings (when supplied)
• CRFs (when supplied)
• Any page with a signature that does not contain other information key to the understanding of the
submission
• Applicants should consider providing signatures on separate pages from key text in reports, overviews, etc.
A2-4 Further Information
If applicants are uncertain whether or not a particular document should be text searchable, they should contact
the relevant NCA for guidance.
Technical eCTD Guidance v4.0 Page 40 of 62

Annex 3: Guidance and Best Practice on the Structure of Module 3
CTD-Quality Considerations for eCTD Submissions in Europe
A3-1 Introduction
The ICH eCTD Specification allows the applicant to manage eCTDs at different levels in Module 3. The normal
choice should be one single eCTD application that covers multiple drug substances, multiple manufacturers,
multiple drug products (components), multiple dosage forms, presentations, invented names and strengths. If
the applicant needs to have one eCTD application per strength or dosage form, this should be explained and
guidance should be given in the cover letter about which documentation differs to prevent duplicate of work
during assessment.
This Annex is based on the use of the ICH eCTD specification v3.2. Refer to the Glossary for an explanation of
terms.
A3-1.1 Attribute Information in the eCTD XML
In addition to CTD-Q documents, eCTD applications provide quality information in XML attributes in the
following locations:
• Module 1: Envelope – INN, Invented Name (Trade Name)
• Leaf: eCTD Title (Submission Description)
• Module 3:
o m3-2-s-drug-substance: substance
o m3-2-s-drug-substance: manufacturer
o m3-2-p–drug-product: product-name
o m3-2-p–drug-product: dosage form
o m3-2-p–drug-product: manufacturer
o m3-2-p-4–control-of-excipients: excipient
o m3-2-a-1-facilities-and-equipment: substance
o m3-2-a-1-facilities-and-equipment: product-name
o m3-2-a-1-facilities-and-equipment: dosage form
o m3-2-a-1-facilities-and-equipment: manufacturer
o m3-2-a-2-adventitious-agent-safety-evaluation: substance
o m3-2-a-2-adventitious-agent-safety-evaluation: product-name
o m3-2-a-2-adventitious-agent-safety-evaluation: dosage form
o m3-2-a-2-adventitious-agent-safety-evaluation: manufacturer
o m3-2-a-3-excipients: excipient
More than 1 entry for each of the Module 3 XML Attributes above generally results in the replication of the
relevant portion of both the XML and the folder architecture, (e.g., 3.2.S Drug Substance, 3.2.P Drug Product,
3.2.P.4 Control of Excipients)
1
.
1
See section A3-3.3.3 Manufacturer ‘Manufacturer of Drug Product’ as an exception, as in some eCTD building
tools only xml is replicated, not the folder structure.
Technical eCTD Guidance v4.0 Page 41 of 62

A3-2 General Principles
A general principle is that the XML index is a reflection of the document granularity, i.e. best practice is to
assign the metadata to agree with the granularity of the CMC dossier rather than designing the granularity
around the metadata.
A3-2.1 Document Granularity
eCTD applications can handle different authoring strategies for CTD-Q information. For any given CTD-Q topic
(e.g., P.1 Description and Composition of the Drug Product), either a single document can be provided that
covers multiple strengths and manufacturers, or multiple documents can be provided, e.g. per strength and/or
per manufacturer. Regardless of the XML attributes, when there are significant differences in content it is best
practice to provide multiple documents, to realise the lifecycle benefit that eCTD offers. When deciding on the
strategy for the single- or multiple-document approach applicants should also take into consideration the ability
of the assessor to review the content. If there are multiple files in the same element, the title of each leaf should
be used to distinguish the scope of each document’s content, and the –var part of the filename used to
differentiate each PDF document.
A3-2.2 Identifying to an Agency What the Application Covers
Generally speaking, multiple eCTD applications can be provided for different strengths and dosage forms.
However, a single eCTD is preferred (see A3-1, Introduction of this annex). A key factor in making this decision
is that in Europe the applicant cannot cross-refer from one eCTD to another (e.g., for drug substance).
A3-2.2.1 Centralised Procedures
For the Centralised Procedure, a single eCTD application should always cover all strengths and dosage forms
within the procedure. A duplicate must always be submitted and maintained as a separate individual eCTD
application.
A3-2.2.2 MR and DC Procedures
In MRP/DCP, a single eCTD is needed per procedure that covers all involved MSs, regardless differences in
the invented name. However, different dosage forms or strengths can be managed in separate eCTDs at the
applicant’s discretion, even if one combined eCTD is preferred. Applicants should carefully consider what an
eCTD application will cover before submitting the first sequence. Refer to Section 2.2
; Structure of Submissions
and Table 16– Advantages and disadvantages of eCTD application structures.
Table 16 (A3): Advantages and disadvantages of eCTD application structures
One Combined eCTD For Multiple Strengths And Dosage Forms
Advantages
Disadvantages
Clinical and non-clinical documentation
is provided only once
Any change to any strength/dosage form will add another
sequence to the application, and therefore the application in
general will eventually contain a larger number of sequences.
Some sequences would cover all products covered by the
eCTD application, other sequences may affect only one
strength or dosage form. Applicants need to use the
submission description to describe what each sequence
covers.
Any changes to drug substance, or
safety related changes that affect the
product, will require only one sequence
Adding a new strength (line extension) could involve
replacing all ‘common’ documents with documents covering
existing strengths plus the new strength, and also adding
new additional strength-specific documents
Common documents can be included
only once (e.g., Pharmaceutical
Development for multiple tablet
strengths)
Lifecycle management becomes more complex in the
following situations:
• In MRP, an applicant applies for only certain strengths, in
certain countries (e.g. strength 1 and 3 in CMS X and
strength 2 and 4 in CMS Y, etc)
• An applicant wants to transfer a certain marketing
Technical eCTD Guidance v4.0 Page 42 of 62

One Combined eCTD For Multiple Strengths And Dosage Forms
authorisation (certain strength) within one eCTD
application to another MAH.
• An applicant wishes to withdraw one strength
• Variations may be only applicable for one specific
strength, and result in the creation of strength specific
documents. These would have to be added to the
lifecycle and managed alongside the existing
documentation, which, if originally ‘common’, would then
only cover the existing (non-affected) strengths
All lifecycle is in one place
Could get complex (e.g., multiple SmPCs)
Documents that are common are
presented only once and therefore read
only once by the assessor
One eCTD Application Per Strength Or Dosage Form
Advantages
Disadvantages
A new strength (line extension) could be
handled in a new eCTD and would not
affect existing lifecycle
All clinical and non-clinical reports are provided for each
strength or dosage form (cannot cross reference across
different eCTDs in the EU)
Lifecycle management can be
maintained per strength so fewer issues
when applying for only certain strengths
in certain countries within MRP/DCP, or
MA-
transfer or withdrawals, line
extensions, variations, etc.
Any changes to the drug substance or changes that affect all
strengths/dosage forms of the product (eg safety related
changes to the labelling) would mean building and submitting
multiple eCTD sequences, one within each eCTD application.
Lifecycle is maintained separately, and would need to be
managed across multiple potentially identical eCTD
applications
If all strengths/dosage forms are not
marketed in every country in an MRP
Procedure, then a unique application per
strength will avoid the possibility that
one CMS will not accept the dossier
because it contains data on a product
which is not being marketed in that
country.
Common documents must be included in each eCTD
application, (cannot cross reference from one eCTD to
another in the EU)
Difficult for the assessor to know what to read/what is unique.
This needs therefore to be thoroughly described in each
submission, which will typically consist of multiple identical
sequences in different eCTD application lifecycles.
This alternative goes against a founding principle for the
management of electronic data insofar as it means :
- loss of storage place: the same information will be archived
several times at different places, sent several time for long-
term filing, and saved several times in the everyday back-up
of servers.
- multiple data entry: data concerning the common part of the
multiple dossiers (i.e. the major part of the dossiers) will have
to be entered several times in the document management
systems, reviewing systems and workflows, both
in NCAs
and in Pharmaceutical companies
At NCAs, uncertainty on whether a MA for a dosage form is
Technical eCTD Guidance v4.0 Page 43 of 62

granted on the basis of assessment of data pertaining to the
dossier of another dosage form
A3-2.2.3 EU Envelope
The Module 1 EU envelope provides the trade name (invented name) of the drug product. The tracking
number element, which is repeatable, may list all of the product licences or application numbers covered by
the eCTD. Applicants should ensure that the values for invented name, INN Applicant and Application Number
in the EU envelope are complete and consistent. Note: The Application Number and INN may not be known at
the time of the first submission and may have to be substituted in later sequences.
A3-3 Module 3 XML Attributes in the eCTD
A3-3.1 Choosing Module 3 XML Attributes
The XML attributes reflect the document granularity used in Module 3. The actual words for the attributes need
not be an exact match of the words used in the content of Module 3 documents. Many eCTD building tool
vendors have based their tools on the ICH style sheet, and this means that the original Module 3 XML
attributes cannot be changed with later submissions within the same application without losing the lifecycle
benefit that eCTD offers. For example, if an applicant builds an eCTD with ABC Chemical as the manufacturer
of substance 1, and subsequently changes supplier to XYZ Chemical, they would normally provide a new
eCTD sequence with XYZ Chemical as an additional module 3 XML attribute. It will not be possible to have
content in the XYZ Chemical section that replaces or appends to content in the ABC Chemical section. This is
because in the eCTD it is not possible to apply lifecycle across different sections. However, it will be possible to
delete some or all of the content within the ABC Chemical section if required.
More than 1 entry for any attribute generally results in the replication of the relevant portion of both the XML
and the folder architecture, (e.g., 3.2.S Drug Substance, 3.2.P Drug Product, 3.2.P.4 Control of Excipients,
3.2.A.1 Facilities and Equipment, 3.2.A.2 Adventitious Agents Safety Evaluation, 3.2.A.3 Excipients).
A3-3.2 Drug Substance (32s) Attributes – Substance-1, Manufacturer-1
The use of these attributes is mandatory. Refer to ICH eCTD Q&As #65 and 66 for guidance.
A3-3.2.1 Drug Substance
The entry for the drug substance name attribute can be an abbreviation of the INN, or if not available, then the
company’s code for the drug substance.
If there is more than 1 drug substance in the application, there is a separate set of 3.2.S.1 to 3.2.S.7 folders
and corresponding XML elements for each drug substance. This also applies for the open (applicant’s) parts of
Active Substance Master Files (ASMFs).
If a drug substance is covered by a Certificate of Suitability (CEP), the certificate is to be provided in
3.2.Regional Information (and in Module 1.2 for annex 5.10). Only relevant sections of its 3.2.S.1 to 3.2.S.7
folders are used, if needed (e.g., for information not covered by the CEP). See EU CTD Q&As Question 12
.
A3-3.2.2 Manufacturer of Drug Substance
In conjunction with the drug substance attribute, each additional manufacturer entry results in additional 3.2.S.1
to 3.2.S.7 XML elements and folders, where there is content provided.
Various approaches are possible depending on the number of manufacturing companies/manufacturing sites
and the quantity of documentation that is manufacturer-specific.
A3-3.2.2.1 Approach 1 – Single XML Section covering all Manufacturers of the Drug Substance
Where drug substance documentation is identical or very similar for all manufacturers (and hence there are a
minimal number of manufacturer-dependent documents), then a non-specific manufacturer attribute can be
used (such as the parent name of a group of companies (but be aware this can also change), or ‘applicant’ or
‘all’). For CTD topics that are manufacturer-specific, having separate documents enables the applicant to
Technical eCTD Guidance v4.0 Page 44 of 62

manage lifecycles as-needed. In such cases, the title and file name of each leaf is to be customised to
differentiate the files, e.g., leaf title of “Batch Analysis - [manufacturer 1]” where the entry for [manufacturer 1] is
either the [current company name] or [current manufacturing town] and file name of batch-analyses-
manufacturer1.pdf. Using leaf titles and filenames to distinguish manufacturers does not involve adding any
extra XML attributes for drug substance manufacturer. As an illustration, see Figure 2, where the specification
is manufacturer-independent but stability data documentation has been separated by manufacturer.
This approach does not prevent a future scenario when a new manufacturer may have its own XML attribute
(due to a significant volume of manufacturer-specific documentation). Note that a known limitation of ICH eCTD
specification v3.2 is that the original, non-specific XML attribute cannot then be modified in the application.
2
2
When any XML attributes is no longer accurate, nor in accordance with this guidance, it is
acceptable to retain original entries. It is not desirable to correct the XML attributes (i.e., applicants need not
apply an operation attribute of DELETE to previously-submitted files and re-submit the latest versions with new
XML attributes).
Technical eCTD Guidance v4.0 Page 45 of 62

Figure 2 (A3) : Single Drug Substance, 2 Manufacturers with similar documentation, the few site/manufacturer-specific documents
are identified by the XML title (not by adding an additional XML section):
XML Files and folders (directory
Arrows indicate destination of xlink:hrefs
Technical eCTD Guidance v3.0 Page 46 of 62

A3-3.2.2.2 Approach 2 – New XML Sections for Each Manufacturer of the Drug Substance
When there are many manufacturer-specific documents, (e.g., if the route of synthesis or
manufacturing process is different per manufacturer), it may be helpful to have additional XML
attributes and equivalent folders for each manufacturer, see Figure 3. Since these files are located in
separate elements, the leaf titles and filenames do not need to be customised per manufacturer. In this
illustration, since the ‘specification’ document is manufacturer-independent, it appears only once in the
folder structure. Additional XML attribute entries are not expected for each intermediate manufacturing
site or packaging site, but can be used.
As an alternative to Approach 1 and Approach 2 (but not illustrated here), an additional entry of
‘common’ may be used for manufacturer-independent documents (e.g., those in 3.2.S.1 General
Information), such that both the XML and the folder structure contain a ‘common’ entry. If this
approach is used, files do not need to be linked from ‘common’ folders to the named manufacturer
folder(s), i.e., these files appear once in the XML and once in the folder directory.
For example, a drug substance section could contain three 32s elements:
32s-aspirin-manufacturer-1 (containing information specific to the manufacturer e.g. 3.2.S.2)
32s-aspirin-manufacturer-2 (containing information specific to the manufacturer e.g. 3.2.S.2)
32s-aspirin-common (containing manufacturer-independent information)
However, this approach can be difficult to review from an assessors perspective, and can lead to
problems later in lifecycle, for example if a third manufacturer is added, and content in the ‘common’
section now only applies to manufacturer 1 and manufacturer 2, and is no longer really ‘common’.
Therefore, this third approach is not recommended.
Technical eCTD Guidance v4.0 Page 47 of 62

Figure 3 (A3) : Single Drug Substance, 2 Manufacturers with significant volume of different documentation (one section for Acme
Chemicals, another for Pharmasupply)
XML Files and Folders (directory)
Technical eCTD Guidance v3.0 Page 48 of 62

A3-3.3 Drug Product (32p) – Product/Dosage Form/Manufacturer
The use of these attributes is optional. Refer to IICH eCTD Q&As #68, 69 and 70 for guidance.
A3-3.3.1 Drug Product Name
Since the M1 EU envelope contains the invented name, it is not necessary to use this name in the
product name XML attribute that is used in Module 3. Applicants should take into consideration that
trade names can occasionally change over time. If the trade name is not well established, applicants
should consider alternatives such as ‘active’, or ‘product’. Alternatively, the internal company code of
the drug product name may be used. If applicable, additional attributes can be used as needed (e.g.
‘diluent’ or ‘placebo’).
This attribute then results in a full set of 3.2.P.1 to 3.2.P.8 XML elements and folders.
A3-3.3.2 Dosage Form
In conjunction with the above product name, each additional dosage form entry results in an additional
full set of 3.2.P.1 to 3.2.P.8 XML elements and folders. When deciding on the degree of detail (e.g.,
‘tablet’ vs. ‘film-coated tablet’, 'frozen" vs. "refrigerated" formulation for vaccines), consider the
potential for future line extensions and the proportion of drug product documents that could be re-used.
If that proportion is small, then consider an initial specific dosage form entry, e.g. ‘film-coated tablet’
rather than ‘tablet’.
Strength(s) need not be mentioned in the attribute. Not all 3.2.P documents are, nor need to be,
strength-dependent. For example, for a common granulation for six strengths, many documents would
have nearly identical content; little benefit would be derived from having strength-specific
documentation. However if there is a chance that some strength(s) may not be approved or may be
later handled in another eCTD application, then some CTD topics might benefit in having separate
leaves per strength (e.g.3.2.P.5.1 Specification).
A3-3.3.3 Manufacturer
If used and in conjunction with the above product name and dosage form, each manufacturer entry
results in a set of 3.2.P.1 to 3.2.P.8, 3.2.A.1 or 3.2.A.2 XML elements. However, in some eCTD
building tools, entries for drug product manufacturer do not result in additional directory folders.
Industry practice is either to not use this attribute or to provide a single high-level descriptor. A general
term such as ‘all’ or ‘applicant’ is acceptable.
If specific manufacturer entries are provided, then the guidance is similar to that for the ‘
Manufacturer
of Drug Substance’. If the building tool did not generate a set of directory folders per manufacturer of
drug product, then corresponding filenames should be customised per manufacturer. Alternatively,
experienced applicants may wish to manually produce a second set of 3.2.P.1 to 3.2.P.8 folders, which
will involve either adding ‘manufacturer’ to the name of the directory folder, (e.g. tablet-5mg-site1), and
editing all xlink:hrefs in the corresponding XML, or editing xlink:hrefs before publishing the eCTD.
Applicants should consult their eCTD tool vendor for further details
.
A3-3.3.3.1 Approach 1 – Single General XML Section Covering All Strengths
If there is a limited number of documents in the submission that are strength-specific, there can be a
single 3.2.P, with a non-specific XML attribute such as ‘tablet’. Where there are multiple files under the
same element, the XML title and file name of each leaf is used to differentiate any documents where
the content is strength-specific, e.g. ‘Stability Data - 100 mg’ and ‘Stability Data - 200 mg’ and ‘stability-
data-100mg.pdf’ and ‘stability-data-200mg.pdf’, respectively. A known limitation of the ICH eCTD
specification v3.2 is that the original, non-specific XML attribute cannot then be modified - see note
under Figure 4.
Figure 4 illustrates this approach, where the Pharmaceutical Development document is strength-
independent but Stability Data documentation has been split by strength.
Technical eCTD Guidance v4.0 Page 49 of 62
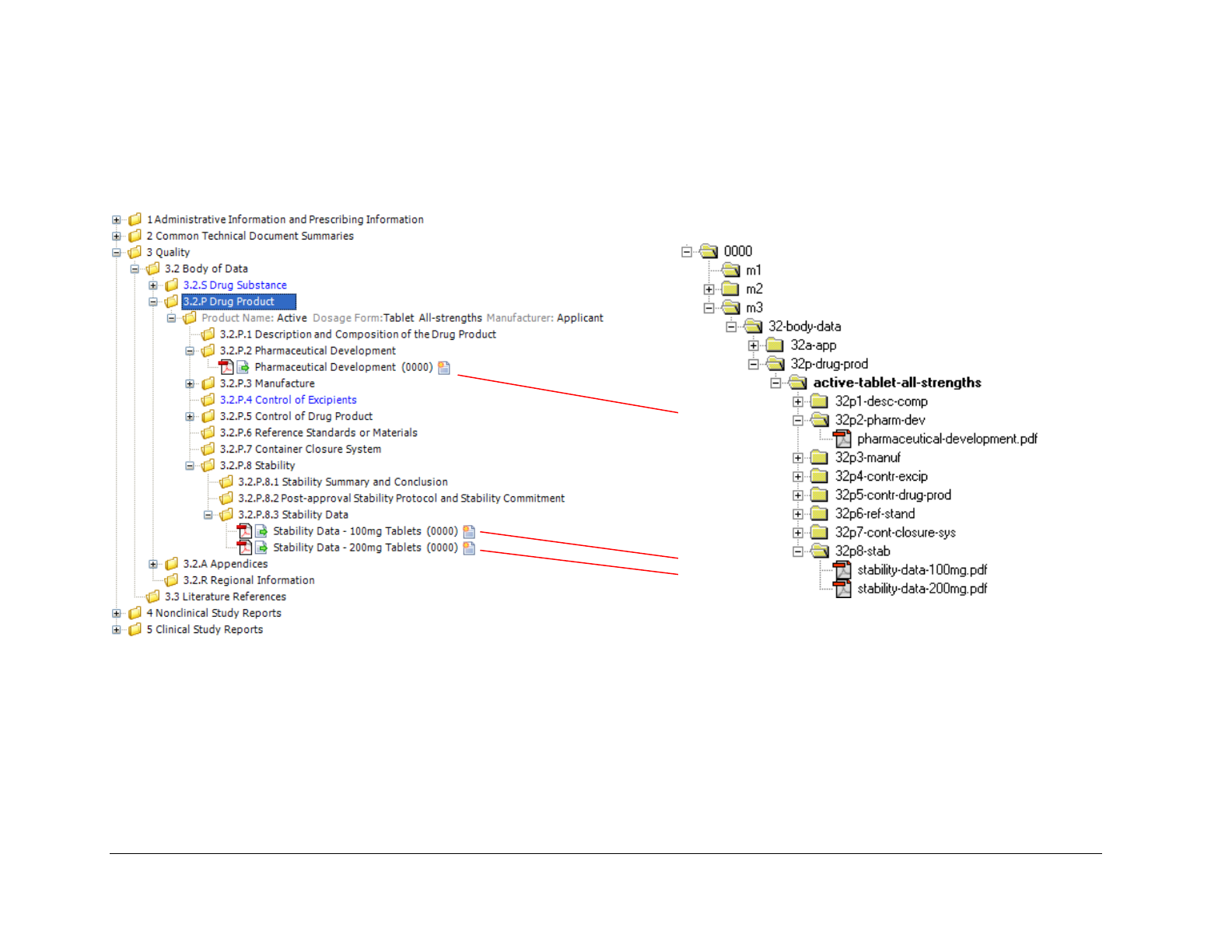
Figure 4 (A3) : Approach 1 – One 32p for all Strengths, any strength specific documents identified by the XML title, not by adding an
additional XML section
XML Files and Folders (directory)
Note: The use of the term ‘all-strengths’ will mean that if the applicant subsequently submits a line extension for an additional strength (e.g.
1000mg) where the documentation is significantly different, and approach 2 is preferred for the new strength, then the attribute ‘all-strengths’
will not include the documentation for the 1000mg tablet. An alternative would be to not use the term ‘all-strengths’ at all and just use ‘Tablet’ for
the dosage form attribute. This implies all strengths and reduces the overall path length.
Technical eCTD Guidance v4.0 Page 50 of 62

A3-3.3.3.2 Approach 2 – Separate XML Section Covering One Strength or Dosage Form
If a strength or dosage form is manufactured in a significantly different way from other
strength(s)/dosage forms and has a large volume of its own 3.2.P documentation, then a separate
3.2.P branch with appropriate subsections applicable to that manufacturer can be justified. In this case,
the dosage form XML attribute and folder name includes the strength (e.g., Tablet 5 mg and tablet-
5mg, respectively). Documentation that pertains to all strengths should only be included once.
Previously-submitted documents or documents that are applicable to more than one strength can be
referred to in new XML leaves under each strength specific XML branch, without re-providing the
content files themselves, see Figure 5.
Technical eCTD Guidance v4.0 Page 51 of 62

Figure 5 (A3) : Approach 2 - Separate XML elements and documents for Strengths – significant content differences, but
Pharmaceutical Development only provided once in the folder structure and referred to from the XML twice
XML Files and Folders (directory)
Technical eCTD Guidance v4.0 Page 52 of 62

A3-3.4 Excipients
The use of these attributes is optional. It is recognised that the current versions of the EU
validation criteria for NeeS and eCTD do not allow for files to be placed directly into 3.2.P.4. This
will be changed the next time the criteria are updated. In the meantime, applicants should note
that the criterion governing file names for eCTD, 15.BP3, is a Best Practice criterion only, and, as
such, applicants are able to use the structures recommended by ICH in Q&A 73. However, for
NeeS, the equivalent criterion (2.11) is a Pass/Fail criterion, and, until the validation criteria are
updated, applicants submitting NeeS will have to adhere to the current EU file naming rules,
which do not allow a single file in 3.2.P.4.
Detailed guidance is provided in the ICH eCTD IWG Question and Answer and Specification
Change Request Document, Q&A #73.
A3-3.4.1 Excipients of Human or Animal Origin and Novel Excipients
Content under sections 3.2.P.4.5 & 3.2.P.4.6 should be provided under an additional attribute
such as ‘animal-human-novel’, refer to ICH eCTD Q&A no. 4. Note, the files provided under this
section should not be in a subfolder to the 32p4-contr-excip folder in the directory structure. Refer
to the ‘File and Folder Structure Names’ worksheet in the eCTD validation rules.
Technical eCTD Guidance v4.0 Page 53 of 62

Annex 4 – Management of Parallel Variations in the eCTD
A4-1 Background
Parallel variations are variations on-going within a single product lifecycle at the same point in
time that are modifying the same content. Tracking these variations and modification of the
content representing the current-approved baseline represents a particular business challenge
within eCTD.
This annex presents the recommended approach for managing parallel variations in accordance
with the ICH and EU eCTD Specifications.
A4-2 Business Challenge
Parallel variations occur when more than one variation is submitted modifying the same approved
content, the first of which has not been approved before the next is submitted. Upon approval of
one of the variations, the approved content has changed. The remaining pending variations
contain proposed content that may be based upon the originally approved content or on the newly
approved content. The applicant must track each separate approval, and update the content for
each pending variation.
The specific challenges associated with this sequence of events are as follows:
(i) Tracking the approved content
(ii) Tracking separate on-going variations
(iii) Tracking individual or combined approvals for each variation
A4-3 Best Practice
Two options are described in this Q&A. Option 1 is the classic approach to eCTD lifecycle
management, where a single document is replaced by a different, updated version, at the initial
submission. Option 2 involves leaving the ‘approved’ content in the eCTD, but introducing
‘proposed’ documents into the lifecycle, until the outcome of the assessment is clear, and only
then replacing the original ‘approved’ content. This Q&A can provide no guidance on when to use
the technique described in option 2, and when to assume that a variation is not happening in
parallel, and therefore to use option 1, submitting the proposed content as a leaf with the ‘replace’
operation attribute. Applicants need to consider whether or not it would be appropriate to use the
one or the other option outlined in this guidance on a case by case basis.
Technical eCTD Guidance v4.0 Page 54 of 62

A4-4 Description of Figures
MAA
0000 (related
sequence – none)
Leaf Title (Operation Attribute)
0001(related
sequence – none)
Leaf Title (Operation Attribute)
eCTD Viewer Current View
0000 Document 1 (New) (0000)
Figure 6 (A4): Description of elements
A4-4.1 Use of one Lifecycle (Option 1)
This option describes the classic eCTD lifecycle management approach, where existing content is
replaced with revised content. When there is only one change and variations do not occur in
parallel, and the change is approved, the advantage of this approach is that applicants do not
have to follow up with another ‘consolidation’ sequence after approval, because they have
already replaced the content and the current view of the eCTD will be correct.
Figure 7 (A4) : Use of one document lifecycle
MAA & Variations
0000 (MAA) (related
sequence – none)
Document 1 (New)
0001 (Variation 1)
(related sequence – none)
Document 1 Proposal 1 (replace)
0002 (Variation 2)
(related sequence – none)
Document 1 Proposal 2 (replace)
eCTD Viewer Current View
0000
Document 1 (New) (0000)
0000 - 0001
Document 1 Proposal 1 (replace) (0001)
0000 - 0002
Document 1 Proposal 2 (replace) (0002)
A4-4.1.1 Drawbacks of Option 1
Approval Status and Clarity on the Basis for Further Changes
If option 1 is used, and the most recently submitted document is replaced, regardless of its
regulatory status, then it becomes unclear what has been approved, and therefore what the
How the sequences would
appear in an eCTD Viewer in
‘current view’
Note – other views may be available,
Sequence numbers, with
related sequence in
b kt
eCTD Leaf Title Attribute and
Operation Attribute in brackets
The red cross indicates the
target of the modified file
Technical eCTD Guidance v4.0 Page 55 of 62

changes in the incoming new submission are based upon. In this example, it is unclear whether
Proposal 2 is based on the content from Proposal 1 or the content from the original Document 1
in sequence 0000. Therefore, when using this approach, if a second variation is submitted before
the first is finalised, applicants should specify which document the changes in the second
variation are based on.
Impact of Regulatory Outcome
If the operation attribute ‘replace’ is used in each variation as described in Figure 1, then
depending on the progress of the review/approval/rejection of each variation, the eCTD lifecycle
may not correctly represent the current or most appropriate lifecycle. For example, if variation 2
were to be approved first, the documentation of variation 1 where changes were based on the
content in sequence 0001 may no longer be displayed appropriately. If variation 1 is rejected, and
variation 2 was based on changes versus the content in variation 1, the content of variation 2
might be difficult to evaluate.
If either variation 1 or 2 (or both) is rejected, a new sequence has to be submitted reflecting only
the approved content.
One advantage of this approach is that if both variations are approved, no additional sequence
has to be submitted.
A4-4.2 Creation of separate Approved and Proposed Document Lifecycles
(Option 2)
This option can be used in any situation where parallel variations are expected, but this is
specifically recommended when submitting labelling changes in the Centralised Procedure, and
the terminology used here is therefore specific to the SmPC in Centralised applications. When an
applicant expects parallel variations to be submitted, the proposed document for the variation is
not submitted as a replacement of the original (approved) content. Instead of using operation
attribute "replace", a new document lifecycle is created for each proposed document by
submitting it with a title identifying its proposed status and a very brief description of the change
being proposed, and an operation attribute of “new”. Proposed content submitted in this way can
only be submitted as a “new” document, or replace other proposed content in the same location.
The approved content in this CTD section has a separate lifecycle, and has a title indicating it is
the approved document. This means that eCTD viewing tools will allow viewing of both the
approved and proposed content alongside each other in the eCTD “current” view. Specifically, for
the m1.3.1 section of the dossier in the Centralised Procedure ‘approved’ documents are labelled
with either ‘Final Opinion’ or ‘Commission Decision’ in the leaf title, depending on the stage of the
review that resulted in the final agreed labelling. Variations are then submitted as ‘new’
documents, not replacing this approved content, with appropriately descriptive leaf titles, for
example, “Type II Variation Section 4.4 Update June 2012 - Proposed”.
A4-4.2.1 Addition of further proposed Document Lifecycles (parallel
variations)
If, during the review of the first variation, there is a subsequent variation that also proposes
changes to the same content, this is also submitted with an operation attribute of “new”, and a
title indicating that the document being provided is another proposal. The document submitted in
the second proposal should reflect changes made versus the current approved document (in this
example, the current decision); the document does not contain the changes from Variation 1. The
leaf title should differentiate it from the former proposed document from the previous variation.
Assigning descriptive title attributes will allow the proposed document in each variation to be
identified by viewing tools displaying the “current” view.
Figure 7 illustrates how the “current” view is able to display each of two parallel variations, when
the title attribute is specific to the variation and the operation attribute “new” is used as described.
Technical eCTD Guidance v4.0 Page 56 of 62

Figure 8 (A4) : Use of separate document lifecycles with different title attributes
Original MAA
Final Opinion or
Decision Only
Variation 1
Variation 2
0000
(rel sequence –
none)
Proposed SmPC
(New)
0001
(rel sequence –
0000)
RTQ 120 changes
to SmPC
(Replace)
0002
(rel sequence –
0000)
RTQ 180 changes
to SmPC
(Replace)
0003
(rel sequence –
0000)
SmPC (English)
Decision Jan
2012 (Replace)
0004
(rel sequence –
none)
Type II Variation
Section 4.4
Update June 2012
- Proposed (New)
0005
(rel sequence –
none)
Type II Variation
Section 4.6
Update July 2012
- Proposed (New)
eCTD Viewer Current View
0000
Proposed SmPC (New) (0000)
0000 - 0001
RTQ 120 changes to SmPC (Replace) (0001)
0000 - 0002
RTQ 180 changes to SmPC (Replace) (0002)
0000 - 0003
SmPC (English) Decision Jan 2012 (Replace) (0003)
0000 - 0004
SmPC (English) Decision Jan 2012 (Replace) (0003)
Type II Variation Section 4.4 Update June 2012 – Proposed (New) (0004)
0000-0005
SmPC (English) Decision Jan 2012 (Replace) (0003)
Type II Variation Section 4.4 Update June 2012 – Proposed (New) (0004)
Type II Variation Section 4.6 Update July 2012 - Proposed (New) (0005)
A4-4.2.2 Upon Approval of a Single Variation – Deletion and Replacement
Upon approval of any variation, the proposed content is deleted, and the original (approved)
content is replaced with a document containing the revised content. Current view will then show
the newly approved content, but also any outstanding proposals, as shown in Figure 8and Figure
9. This additional sequence should be considered part of the same regulatory activity as the
proposal that has been approved.
Figure 9 (A4) : Replacement of approved content by newly approved content, and deletion
of proposed content
In this example, commission decision received in December 2012 incorporates the changes from
the Type II variation submitted in sequence 0004 only. The changes from the Type II variation in
sequence 0005 are still under review. In sequence 0006, the ‘proposed’ label from the first
variation is deleted and the new commission decision is provided, replacing the original
commission decision from sequence 0003. The content of the second variation is also amended
in sequence 0006, to reflect the newly approved content in section 4.4, alongside the still
unapproved change in section 4.6, and left in the lifecycle as an outstanding proposal.
Technical eCTD Guidance v4.0 Page 57 of 62

MAA
Final Opinion or
Decision Only
Variation 1
Variation 2
0003
(rel sequence –
0000)
SmPC (English)
Decision Jan 2012
(Replace)
0004
(rel sequence –
none)
Type II Variation
Section 4.4 Update
June 2012 -
Proposed (New)
0005
(related sequence –
none)
Type II Variation
Section 4.6 Update
July 2012 -
Proposed (New)
0006
(rel sequence –
0004, 0005)
SmPC (English)
Decision Dec
2012 (Replace)
Type II Variation
Section 4.4 Update
June 2012 -
Proposed (Delete)
Type II Variation
Section 4.6
+ inclusion of
approved Type II
Variation Section
4.4
Update Dec 2012 –
Proposed (Replace)
eCTD Viewer Current View
0000 - 0005
SmPC (English) Decision Jan 2012 (Replace) (0003)
Type II Variation Section 4.4 Update June 2012 - Proposed (New) (0004)
Type II Variation Section 4.6 Update July 2012 - Proposed (New) (0005)
0000 - 0006
SmPC (English) Decision Dec 2012 (Replace) (0006)
Type II Variation Section 4.6 Update Dec 2012 - Proposed (Replace)
(0006)
Technical eCTD Guidance v4.0 Page 58 of 62

Figure 10 (A4) : Progression on the 2nd Parallel Proposal
In this example, as the procedure progresses, the proposed change to section 4.6 needs to be
further amended as a result of the regulatory review (e.g. updated wording as suggested by the
assessor), and this amendment is done with a replacement document in sequence 0007. Once a
decision is reached, another sequence, 0008, is submitted, including the new decision replacing
the one submitted in sequence 0006, and a deletion of the section 4.6 proposal, as amended by
sequence 0007.
MAA
Final Opinion or
Decision Only
Variation 1
Variation 2
0003
(rel sequence
– 0000)
SmPC (English)
Decision Jan 2012
(Replace)
0004
(relsequence –
none)
Type II Variation
Section 4.4 Update
June 2012 -
Proposed (New)
0005
(rel sequence
– none)
Type II Variation
Section 4.6 Update
July 2012 - Proposed
(New)
0006
(rel sequence
– 0004, 0005)
SmPC (English)
Decision Dec 2012
(Replace)
Type II Variation
Section 4.4 Update
June 2012 -
Proposed (Delete)
Type II Variation
Section 4.6
+ inclusion of
approved Type II
Variation Section 4.4
Update Dec 2012 –
Proposed (Replace)
0007
(rel sequence
– 0005)
Type II Variation
Section 4.6 +
inclusion of
approved Type II
Variation Section 4.4
Update January
2013 – Proposed
(Replace)
0008
(rel sequence
– 0005)
SmPC (English)
Decision
Mar 2013
(Replace)
Type II Variation
Section 4.6 +
inclusion of
approved Type II
Variation Section 4.4
Update January
2013 – Proposed
(Delete)
eCTD Viewer Current View
0000 - 0006
SmPC (English) Decision Dec 2012 (Replace) (0006)
Type II Variation Section 4.6 Update Dec 2012 - Proposed (Replace) (0006)
0000 - 0007
SmPC (English) Decision Dec 2012 (Replace) (0006)
Type II Variation Section 4.6 Update Jan 2013 - Proposed (Replace) (0007)
0000-0008
SmPC (English) Decision Mar 2013 (Replace) (0008)
A4-4.2.3 Approval of Multiple Variations at the Same Time
There will be occasions where multiple changes are approved at the same time, or within a very
short time period. In these instances, it would not be appropriate to submit separate ‘approved’
Technical eCTD Guidance v4.0 Page 59 of 62

content for each variation; instead, a consolidated document representing all the approved
changes should be submitted. This would replace the existing ‘approved’ content. The eCTD
sequence containing the new approved content would also contain leaves deleting the relevant
‘proposed’ documents, as illustrated in Figure 5.
Figure 11 (A4) : Replacement of approved content by newly approved content, and
deletion of multiple proposed documents
In this example, commission decision received in December 2012 incorporates the changes from
the Type II variation submitted in sequence 0004, and also the changes from the Type II variation
in sequence 0005. In sequence 0006, both ‘proposed’ documents from the variations are deleted
and the new commission decision is provided, replacing the original commission decision from
sequence 0003. This leaves only the latest commission decision from sequence 0006 in the
current view.
MAA
Final Opinion
or Decision
Only
Variation 1
Variation 2
0003
(related sequence
– 0000)
SmPC (English)
Decision Jan
2012 (Replace)
0004
(related sequence
– none)
Type II Variation
Section 4.4
Update June 2012
- Proposed (New)
0005
(related sequence
– none)
Type II Variation
Section 4.6 Update
July 2012 - Proposed
(New)
0006
(related sequence
– 0004 & 0005)
SmPC (English)
Decision Dec
2012 (Replace)
Type II Variation
Section 4.4
Update June 2012
- Proposed
(Delete)
Type II Variation
Section 4.6 Update
July 2012 - Proposed
(Delete)
eCTD Viewer Current View
0000 - 0005
SmPC (English) Decision Jan 2012 (Replace) (0003)
Type II Variation Section 4.4 Update June 2012 - Proposed (New)
Type II Variation Section 4.6 Update July 2012 - Proposed (New)
0000 - 0006
SmPC (English) Decision Dec 2012 (Replace)
A4-4.2.4 Rejections or Withdrawals
If any proposed changes are rejected or withdrawn by the applicant, then the applicant can
provide a sequence deleting the “proposed” content document, but not providing any replacement
for the original approved document.
A4-4.2.5 Typical Applications
An applicant is most likely to need to submit parallel variations on dossier content that changes
more often, such as the SmPC or the Risk Management Plan (RMP
i
). However, this guidance is
not limited to use in these parts of the dossier. Applicants should also note that many variations
affecting the SmPC and RMP will not occur in parallel with another variation, and at times, when
a variation is initially expected to be completed in isolation, a second parallel variation may
become necessary at a later stage.
Technical eCTD Guidance v4.0 Page 60 of 62

Document Control
Change Record
Version
Author(s)
Comments
1.0
TIGes eCTD guidance
topic group
This document has been prepared by the eCTD Guidance Topic Group of the
TIGes. It is largely based on the NeeS guidance document 1.4.
1.1
GW, AN, KG, KM
First draft for revision, made the document in line with agreed text in the
NeeS guidance and with the New validation criteria, TIGes Harmonisation
group 110309
1.2-1.93
TIGes Harmonisation
Group/ AN, KG, MC,
KM, BT, KP
Further revisions by TIGes Harmonisation group TC meetings in April-July
2011
1.94
KG
Further revision after TIGes comments and minor update in accordance with
EU M1 eCTD specification draft v.1.4.1.
2.0
KG, AN
Following review of TIGes comments and final updates at subgroup TC
meeting in August. Final document for TIGes adoption and publication.
2.x
KG, AN
Draft for Second revision, made the document in line with agreed text on
Q&A, EU M1 Spec v2.0, and in the NeeS guidance and with the updated
validation criteria, TIGes Harmonisation group 12/2012-07/2013
2.7
AN, KM
Following review comments edits have been included.
3.1
AN, KM, KG, KP
Preparing the revision to accommodate regulatory changes, improvements and
clarifications, editions due to inconsistencies caused by revisions of the EU
M1 spec. v3.0, eCTD validation criteria 6.0, eAF usage and further
modifications
3.2
KM
Consolidating all changes and formatting modifications
3.3
HHMG
Reviewing modified sections and tiding up comments
3.4
KM
Preparation for publication
Reviewers
Version
Name
Organisation
1.0
TIGes
TIGes eCTD Topic Group
1.1-1.92
Members of the subgroup
TIGes Harmonisation group
1.93
Members of TIGes
TIGes
1.94
Members of the subgroup
TIGes Harmonisation group
2.0
Members of subgroup And the TIGes
TIGes
2.6
Members of the TIGes
TIGes
3.1
Members of HHMG
Human Harmonisation Maintenance Group
3.2
Members of EMA
EMA
3.3
HHMG / eSub CMB
Human Harmonisation Maintenance Group / eSubmission
Change Management Board
Distribution
Version
Distributed to
Way of distribution
1.0
General public
Published on the EMA eSubmission website
1.1-1.92
Members of the Subgroup
E-mail April-July 2011
1.93
Members of the Subgroup and
Members of TIGes
E-mail July 2011
1.94
Members of the Subgroup
E-mail August 2011
2.0
Members of the Subgroup and
Members of TIGes
E-mail August 2011
3.0
Members of TIGes
E-mail August 2013 /
3.1
eSubmission CMB
E-mail December 2015
3.4
eSubmission CMB
E-mail March 2016
Coming into Operation
Version
Date in operation
Comment
1.0
May 2009
This document is specifically called a “Draft for Testing”. The Topic Group
Technical eCTD Guidance v4.0 Page 61 of 62

fully anticipate comments from NCAs and applicants which will enable future
versions to reflect practical experience of users. In this way the document will
evolve to become an essential work of reference in this area.
2.0
1 September 2011
Published at the EMA eSubmission website for use
3.0
2 September 2013
Published at the EMA eSubmission website for use
4.0
01. July 2016
Published at the EMA eSubmission website for use
Technical eCTD Guidance v4.0 Page 62 of 62
